

Fetal growth restriction in the rare co-occurrence of chromosomal and monogenic diseases
Bolshakova A.S., Barkov I.Yu., Frankevich N.A., Yarygina T.A., Shmakov R.G.
In this article, we report a unique case of simultaneous occurrence of trisomy X and incontinentia pigmenti (IP) in a prematurely born girl with extremely low birth weight, a scenario that has not been documented in literature worldwide.
Understanding fetal growth restriction (FGR) is of paramount importance, as it is a major cause of stillbirth, neonatal morbidity, and mortality. Recent studies have reported the efficacy of noninvasive prenatal screening (NIPS) for fetal aneuploidies through maternal blood sampling as a valuable tool in the management of high-risk pregnancies. This method enables the detection of major fetal aneuploidies, including numerical abnormalities in the sex chromosomes.
In our clinical observation, based on the results of early combined screening, the patient was identified as at high risk for preeclampsia and FGR, both of which, despite preventive measures, occurred at the end of the second trimester of pregnancy. From the first days of life, the newborn girl developed characteristic skin lesions associated with IP, inherited in an X-linked manner. Subsequent chromosomal analysis revealed abnormal 47,ХХХ karyotype in the child.
Conclusion: This clinical observation demonstrates the effectiveness of contemporary ante- and postnatal diagnostic methods for identifying rare combined genetic pathologies associated with FGR.
Authors' contributions: Bolshakova A.S., Barkov I.Yu., Frankevich N.A., Yarygina T.A., Shmakov R.G. – conception and design of the study; Bolshakova A.S., Yarygina T.A., Frankevich N.A. – drafting of the manuscript; Barkov I.Yu., Frankevich N.A., Shmakov R.G. – data collection and processing; Shmakov R.G. – editing of the manuscript.
Conflicts of interest: The authors have no conflicts of interest to declare.
Funding: The study was conducted within the framework of State Assignment № 121040600408-4: "Improving the management and timing of delivery of pregnant women with fetal growth restriction based on the study of molecular genetics and metabolic factors, followed by the introduction of modern methods for diagnosing the severity of this complication of gestation".
Ethical Approval: The study was reviewed and approved by the Research Ethics Committee of the V.I. Kulakov NMRC for OG&P.
Patient Consent for Publication: The patient provided an informed consent for the publication of her data and associated images.
Authors' Data Sharing Statement: The data supporting the findings of this study are available upon request from the corresponding author after approval from the principal investigator.
For citation: Bolshakova A.S., Barkov I.Yu., Frankevich N.A., Yarygina T.A., Shmakov R.G. Fetal growth restriction in the rare co-occurrence of chromosomal and monogenic diseases.
Akusherstvo i Ginekologiya/Obstetrics and Gynecology. 2024; (1): 74-81 (in Russian)
https://dx.doi.org/10.18565/aig.2023.288
Keywords
fetal growth restriction
extremely low birth weight
non-invasive prenatal screening for aneuploidies
trisomy X
incontinentia pigmenti (or Bloch–Sulzberger syndrome)
Fetal growth restriction (FGR), along with pre-eclampsia and preterm birth, is one of the so-called "major obstetric syndromes" [1]. These conditions are associated with abnormal placentation due to remodeling defects and obstructive lesions of the spiral arteries, and are major contributors to perinatal and maternal morbidity and mortality [2].
Recognizing the significance of these conditions, population-based combined screening of the first trimester of pregnancy has been introduced in the Russian Federation since 2021 [3]. This screening aims to detect structural malformations, high-risk chromosomal abnormalities, FGR, and cases with a high risk of preterm birth and preeclampsia [4].
FGR can occur as a result of the influence of isolated or combined pathological maternal and placental factors, as well as genetic and structural abnormalities of the fetus itself, which are detected much more often than in normally growing fetuses [5–8].
Modern criteria for antenatal diagnosis of FGR, approved in the 2016 Delphi procedure [9] and recommended for universal use by the Practice Guidelines of the International Society of Ultrasound in Obstetrics and Gynecology (ISUOG, 2020) [10], place special emphasis on the absence of congenital malformations of the fetus for cases in which such a classification should be applied and approved by the Ministry of Health of the Russian Federation [11].
However, even in fetuses without structural malformations but with growth restriction according to prenatal ultrasound, there is an increased frequency of genetic pathology compared to normally growing fetuses [7, 8].
Considering the possibility of such an association when identifying fetuses with growth restriction, especially its early form diagnosed before the 32nd week of pregnancy [11, 12], the expectant mother should consult a geneticist, discuss possible options, and select genetic testing methods [11, 13, 14].
Modern genetic diagnostic methods enable the identification of chromosomal, submicroscopic, and monogenic disorders that may cause FGR [6]. Non-invasive prenatal screening (NIPS) and invasive prenatal diagnostics, such as chorionic villus sampling and amniocentesis, are currently used to identify genetic abnormalities in the fetus [6, 15]. NIPS is a highly sensitive and specific screening method for trisomies 21, 18, and 13 as well as sex chromosome aneuploidies [16, 17], the latter being the most common in live births.
Despite the current possibilities of instrumental and laboratory methods for examination of pregnant women, the uniqueness of individual clinical cases allows for the completion of the diagnostic search only in the postnatal period.
Clinical observation
A 30-year-old patient was referred to the V.I. Kulakov NMRC for OG&P of the Russian Ministry of Health (hereinafter referred to as the Center) to monitor the course of her pregnancy. From anamnesis, it is known that in childhood, the patient suffered from chickenpox and rubella. At the age of 16 years, the patient had a single convulsive attack, was diagnosed with epilepsy, and was treated with valproic acid for one year; at the time of pregnancy, the patient was removed from the neurologist’s outpatient register. The gynecological history is complicated by hematocolpos from the time of menarche due to complete fusion of the hymen, which required surgical defloration.
This was the first pregnancy that occurred as a result of natural conception in a registered marriage. In the first trimester, the pregnancy was complicated by moderate vomiting, and outpatient treatment was performed.
During prenatal screening at 12 weeks of pregnancy, ultrasound examination revealed no signs of congenital malformations or chromosomal abnormalities in the fetus. At the same time, the pulsatility index in the uterine arteries exceeded the median values [4] and corresponded to 1.4 values that are multiples of the medians (MoM). The mean arterial pressure also increased, amounting to 99.2 mm Hg, which corresponds to 1.16 MoM. In contrast, placental hormone levels in the biochemical screen were below median values, corresponding to 0.34 MoM for free beta subunit of human chorionic gonadotropin, 0.83 MoM for pregnancy-associated protein A, and 0.58 MoM for placental growth factor. According to the combined screening data, a low risk of trisomy of chromosome 21 (1:11333), 18, and 13 in the fetus (<1 in 20000), a high risk of FGR (1:97), and pre-eclampsia (1:24) was established; anti-aggregant prophylaxis of these complications with low-dose aspirin was initiated [3].
To increase the sensitivity of prenatal detection of aneuploidy in the fetus at 14 weeks, the patient underwent NIPS [18], which revealed a high risk of sex chromosome disorders in the female fetus. The geneticist recommended invasive prenatal diagnosis [19], which the woman refused, and decided to prolong the pregnancy regardless of the possibility of fetal karyotype abnormalities.
Starting at 19 weeks of pregnancy, the results of dynamic Doppler studies of uteroplacental blood flow demonstrated persistent disturbances with a pronounced increase in the values of the pulsatility index in the uterine arteries, reaching the hundredth percentile and without a tendency to decrease. Fetal dimensions did not exceed the reference intervals until the 25th week of pregnancy, when early fetal growth restriction was diagnosed, and the estimated weight corresponded to the 2nd percentile.
At 24 weeks of pregnancy, the patient was hospitalized in the obstetric department of the Center. Antihypertensive and anticoagulant therapy was started in connection with the appearance of gestational arterial hypertension in the context of a high risk of preeclampsia. Despite the entire range of preventive and therapeutic measures, at 28–29 weeks of pregnancy, due to the rapid increase in the severity of preeclampsia (increasing edema against the background of a decrease in the rate of diuresis, unstable blood pressure with an increase to 170/110 mmHg during multicomponent antihypertensive therapy), the patient was transferred to the intensive care unit for treatment and stabilization of her condition. However, due to a further increase in the severity of preeclampsia, an increase in edema with generalization, and the addition of ascites and effusion into the pleural cavities at 29 weeks 4 days, delivery was performed by emergency cesarean section.
A live preterm girl was born with Apgar scores of 6 at 1 min and 7 at 5 minutes, birth weight of 900 g (5.3 percentile, − 1.6 Z-score), and body length of 32 cm (0.4 percentile, -2.6 Z-score) [20], which meets the 2018 Delphi consensus criteria for fetal growth restriction [21].
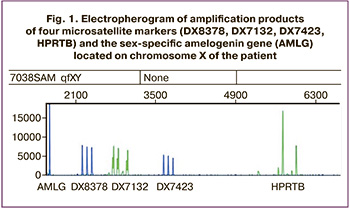
Because of the high risk of trisomy X based on NIPS, chromosomal analysis was performed, which showed the 47, XXX karyotype. Using quantitative fluorescence polymerase chain reaction, the maternal origin of trisomy X was determined by comparing short tandem repeat (STR) loci. The girl was diagnosed with trisomy on chromosome X (XXX) for all the markers (Fig. 1).
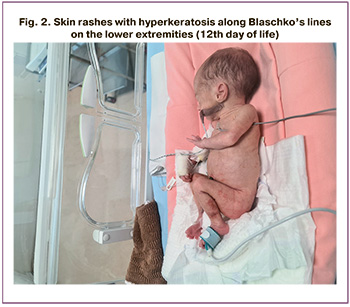
From the first day of life, the child had multiple elements of vesiculopustular rash on the limbs, trunk, and back (Fig. 2); therefore, local therapy included lotions based on calamine, zinc oxide, and dexpanthenol. On the 12th day of life, in addition to vesicles, warty rashes with hyperkeratosis were observed along Blaschko’s lines. No eye damage was observed.
Upon repeated careful history taking, similar skin lesions were observed in the mother after birth. They almost completely disappeared at approximately 6 months of age. However, currently, areas of depigmentation remain in the lower extremities.
This allowed us to suspect IP or Bloch–Sulzberger syndrome in the child [22]. To confirm this, a molecular genetic study was performed.
Genomic DNA was extracted from peripheral blood leukocytes using the PREP-MB MAX DNA isolation kit (Russia).
Analysis by determining the length polymorphism of amplified fragments was carried out for a common deletion of the IKBKG gene (NEMO) and included a polymerase chain reaction (PCR) followed by detection of amplification products by electrophoresis based on the difference in the lengths of normal/mutation fragments.
Considering the clinical picture, family history, and molecular genetic examination findings, the child was diagnosed with vesicular stage of IP (or Bloch–Sulzberger syndrome). No pathological neurological symptoms were noted upon examination by a neurologist. Neurosonography of the brain revealed no focal changes. Electroencephalography on the 8th day of life revealed a normal picture. Audiological screening has been passed; the child is not classified as being at a high risk for developing hearing impairment. The neonatal period proceeded in accordance with the postconceptual age. Comprehensive therapy included infusion, antibacterial, antifungal, antianemic, eubiotic agents, and vitamins, as well as prevention of rickets, bronchopulmonary dysplasia, and stimulation of the respiratory center. The child was discharged in a satisfactory condition with a weight of 2100 g on the 54th day of life.
The girl's mother was advised on proper care and timely examination of the child by relevant specialists within the recommended timeframe. The patient was advised to undergo genetic examination followed by pre-pregnancy monitoring by an obstetrician-gynecologist and therapist at the stage of planning a future pregnancy.
Discussion
Fetal growth restriction can be caused by maternal, placental, fetal, or genetic factors [23].
According to the first data published several decades ago, fetal chromosomal abnormalities account for up to 19% of the total number of births with FGR [24].
However, at the current level of early antenatal diagnosis and elimination of fetuses with structural defects and/or syndromic pathology, an abnormal karyotype is detected in approximately 4% of fetuses with fetal growth restriction [8].
Until recently, fetal cells for karyotyping were obtained only through invasive manipulation. The introduction of NIPS into practice, in addition to biochemical and sonographic markers, can help reduce the number of intrauterine manipulations, and consequently, the complications associated with them [18, 25].
Although screening for sex chromosome aneuploidies using NIPS has more limitations than the detection of trisomies 21, 18, and 13, it demonstrates high sensitivity and specificity along with a moderate positive predictive value [26].
In the presented clinical observation of early FGR, NIPS made it possible to suspect an abnormality of the sex chromosomes, which was confirmed by a postnatal study that revealed trisomy of chromosome X.
Trisomy X is the presence of an additional X chromosome in women (47, XXX instead of 46, XX), occurring in approximately one case per 1000 girls born [17]. Because there are variable phenotypic presentations, ranging from asymptomatic and very mild lesions to significant physical and psychological features, the population incidence of this disorder is thought to be highly imprecise, perhaps because only 10% of people with trisomy X are aware of their diagnosis [27, 28].
Trisomy X occurs due to non-disjunction in the cell division cycle during gametogenesis or after conception. Studies have shown the predominance of maternal gametes; only 10% of cases are associated with paternal mutations [29].
Although karyotype 47, XXX is most often an incidental finding during NIPS [30], cases of a combination of this type of aneuploidy with fetal growth restriction have been described [8].
A number of authors have also noted that newborn girls with trisomy X have lower birth weights and smaller head circumferences than healthy children [31].
In the clinical observations we described, this abnormal karyotype of the child could contribute to the development of FGR. The additional negative effect of the combination of this pathology with Bloch–Sulzberger syndrome cannot be excluded.
IP or Bloch–Sulzberger syndrome is a rare X-linked dominant genodermatosis with a prevalence of approximately 0.7/100,000 live births [32, 33].
IP is caused by a mutation in the IKBKG gene located in the chromosomal region Xq28, which encodes the NEMO protein. Most patients with PI have a deletion of exon 4–10 in the IKBKG gene [34–39].
Interestingly, 65–75% of cases of the disease are caused by sporadic mutations, and the rest are familial.
The disease occurs predominantly in women because the mutation is lethal for male fetuses, leading to antenatal death even before the start of the second trimester of pregnancy. In rare cases, male fetuses with a combination of PI and somatic mosaicism or Klinefelter syndrome may survive. In such situations, paternal transmission of the mutation may occur [32, 33, 40–42].
IP is a multisystem disease affecting the skin, eyes, hair, central nervous system and teeth [22, 32, 33, 43, 44], which requires regular multidisciplinary monitoring of affected children and adults.
The clinical picture of IP varies widely, even among close relatives with a familial pattern of inheritance [36]. The key to IP diagnosing, as in the described case, is specific skin manifestations, which can develop classically in four stages: (1) erythema and vesico-pustules, (2) hyperkeratotic verrucous papules and plaques, (3) hyperpigmented spots, and (4) hypopigmented atrophic plaques with alopecia and occur simultaneously [37, 38]. For example, in the presented clinical observation, at the time of discharge, the child had overlapping signs of vesiculobullous and verrucous stages.
Isolated cases of postnatal diagnosis of PI have been published in the domestic and foreign literature, and although in most of them, the children had a weight corresponding to the gestational age [45–52], in one case of inheritance of a mutation from the father, a girl with PI was born at 36–37 weeks of pregnancy with a weight of 2079 g (-1.3 SD) and a height of 43.5 cm (-1.4 SD) [40].
Of particular note is the case of a patient with pigmentary incontinence, whose pregnancy was complicated by antenatally diagnosed, postnatally pathomorphologically confirmed placental dysfunction, and ended in labor at 36 weeks of pregnancy due to premature rupture of amniotic fluid. In a girl weighing 2640 g, 47 cm in height, born with moderately severe neurological symptoms due to intrauterine hypoxia, with the presence of many blisters with transparent contents on the skin, the diagnosis of IP was subsequently confirmed [53].
In our clinical observation, there is a high probability of a familial form of inheritance of IP from a mother who has had characteristic skin manifestations since birth, as well as an anamnesis aggravated by the pathology of the nervous and reproductive systems, complicated by the course of pregnancy.
It is important to note that patients with IP of reproductive age are characterized by a high frequency of spontaneous abortions, caused both by the death of male fetuses with the mutation and by the frequency of various aneuploidies detected in 50–70% of embryos during preimplantation genetic testing in assisted reproductive program technologies [54, 55].
To date, only one report of a living child with a combination of IP and aneuploidy, trisomy 21, has been published [56].
To date, there have been no reports of a diagnosed combination of IP and trisomy X in children with antenatal growth restriction; therefore, predicting long-term clinical manifestations is difficult.
Normally, women experience random inactivation of an X chromosome. Most women with IP experience non-random inactivation of one X chromosome in the blood and fibroblasts due to the loss of cells with the mutation [57].
The presence of a supernumerary X chromosome may lead to an imbalance in the number of genes that escape inactivation. Therefore, it is possible that our patient had milder clinical manifestations of IP; however, this effect may have been limited.
Women with trisomy X are more likely to experience delayed menarche and reduced fertility [58], but the opposite has also been reported [59].
Hormonal examinations of such patients often indicate ovarian failure and an increased risk of autoimmune diseases, including those affecting the thyroid gland; therefore, careful monitoring by gynecologists and endocrinologists is necessary.
In addition, data have been published on the connection between trisomy X and disorders of cerebral structures and pathological abnormalities in higher nervous system activity [60–62].
It is important to note that FGR and prematurity with extremely low birth weight, even when isolated from genetic pathologies, are high-risk factors for the development of early and late cardiovascular, neurological, ophthalmological, and endocrine complications in the child, leading to a decrease in the quality and shortening of the duration of the upcoming pregnancy life [63–65].
For the comprehensive care of our mother-child patients, long-term and close interdisciplinary collaboration between obstetricians-gynecologists, dermatologists, pediatricians, neurologists, endocrinologists, and geneticists is critical [66–68].
Conclusion
In this clinical observation, which has not been previously reported in the literature, the combination of trisomy X and IP in a child with prenatally developed early growth restriction provides insight into the capabilities and limitations of modern invasive and noninvasive methods for ante- and postnatal diagnosis of combined genetic pathologies.
The intricate and possibly unpredictable interplay of genetic and perinatal factors highlights the essential need for interdisciplinary collaboration between medical and pedagogical support systems to ensure the well-being of children throughout their lives.
Timely diagnosis of genetic pathology in fetuses, children, and adult patients is crucial for delivering specialized medical care and reducing morbidity, disability, and mortality rates both nationally and globally.
References
- Brosens I., Pijnenborg R., Vercruysse L., Romero R. The «great obstetrical syndromes» are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 2011;204(3):193-201. https://dx.doi.org/10.1016/j.ajog.2010.08.009.
- Соколовская Т.А., Ступак В.С. Заболеваемость беременных женщин в Российской Федерации: тенденции и прогнозы. Российский вестник акушера-гинеколога. 2022;22(5):7 14. [Sokolovskaya T.A., Stupak V.S. Morbidity of pregnant women in the Russian Federation: trends and prognosis. Russian Bulletin of Obstetrician-Gynecologist. 2022;22(5):7 14. (in Russian)]https://dx.doi.org/10.17116/rosakush2022220517.
- Министерство здравоохранения Российской Федерации. Приказ от 20.10.2020 №1130н «Об утверждении Порядка оказания медицинской помощи по профилю «акушерство и гинекология». Приложение 9. Зарегистрировано в Минюсте России 12 ноября 2020 г. №60869. [Ministry of Health of the Russian Federation. Order No. 1130n dated October 20, 2020 «On approval of the Procedure for the provision of medical care in the field of obstetrics and gynecology». Supplement 9. Registered with the Ministry of Justice of Russia on November 12, 2020 No. 60869. (in Russian)].
- Ярыгина Т.А., Батаева Р.С. Методика проведения скринингового исследования в первом триместре беременности с расчетом риска развития преэклампсии и задержки роста плода по алгоритму Фонда медицины плода (Fetal Medicine Foundation). Ультразвуковая и функциональная диагностика. 2018;4:77-88. [Yarygina T.A., Bataeva R.S. Methodology of 1st trimester screening for preeclampsia and intrauterine growth restriction according to Fetal Medicine Foundation algorithm (FMF). Ultrasound and Functional Diagnostics. 2018;4:77-88. (in Russian)].
- Brodsky D., Christou H. Current concepts in intrauterine growth restriction. J. Intensive Care Med. 2004;19(6):307-19. https://dx.doi.org/10.1177/0885066604269663.
- Meler E., Sisterna S., Borrell A. Genetic syndromes associated with isolated fetal growth restriction. Prenat. Diagn. 2020;40(4):432-46.https://dx.doi.org/10.1002/pd.5635.
- Wang L.Q., Fernandez-Boyano I., Robinson W.P. Genetic variation in placental insufficiency: What have we learned over time? Front. Cell Dev. Biol. 2022;10:1038358. https://dx.doi.org/10.3389/fcell.2022.1038358.
- Wu X., He S., Li Y., Guo D., Chen X., Liang B. et al. Fetal genetic findings by chromosomal microarray analysis and karyotyping for fetal growth restriction without structural malformations at a territory referral center: 10-year experience. BMC Pregnancy Childbirth. 2023;23(1):73. https://dx.doi.org/10.1186/s12884-023-05394-y.
- Gordijn S.J., Beune I.M., Thilaganathan B., Papageorghiou A., Baschat A.A.,Baker P.N. et al. Consensus definition of fetal growth restriction: a Delphi procedure. Ultrasound Obstet. Gynecol. 2016;48(3):333-9.https://dx.doi.org/10.1002/uog.15884.
- Lees C.C., Stampalija T., Baschat A., da Silva Costa F., Ferrazzi E., Figueras F. et al. ISUOG practice guidelines: diagnosis and management of small-for-gestational-age fetus and fetal growth restriction. Ultrasound Obstet. Gynecol. 2020;56(2):298-312. https://dx.doi.org/10.1002/uog.22134.
- Министерство здравоохранения Российской Федерации. Клинические рекомендации «Недостаточный рост плода, требующий предоставления медицинской помощи матери (задержка роста плода)». 2022. [Ministry of Health of the Russian Federation. Clinical recommendations «The insufficient growth of the fetus, requiring the provision of medical care of the mother (fetal growth)». 2022. (in Russian)].
- Ярыгина Т.А., Гус А.И. Задержка (замедление) роста плода: все, что необходимо знать практикующему врачу. Акушерство и гинекология. 2020;12:14-24. [Yarygina T.A., Gus A.I. Fetal growth restriction (retardation): everything the practitioner should know. Obstetrics and Gynecology. 2020;(12):14-24 (in Russian)]. https://dx.doi.org/10.18565/aig.2020.12.14-24.
- Fetal Growth Restriction: ACOG Practice Bulletin, Number 227. Obstet. Gynecol. 2021;137(2):e16-e28. https://dx.doi.org/10.1097/AOG.0000000000004251.
- Kehl S., Dötsch J., Hecher K., Schlembach D., Schmitz D., Stepan H., Gembruch U. Intrauterine growth restriction. Guideline of the German Society of Gynecology and Obstetrics (S2k-Level, AWMF Registry No. 015/080, October 2016). Geburtshilfe Frauenheilkd. 2017;77(11):1157-73.https://dx.doi.org/10.1055/s-0043-118908.
- Merriel A., Alberry M., Abdel-Fattah S. Implications of non-invasive prenatal testing for identifying and managing high-risk pregnancies. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021;256:32-9. https://dx.doi.org/10.1016/j.ejogrb.2020.10.042.
- Carlson L.M., Vora N.L. Prenatal diagnosis: screening and diagnostic tools. Obstet. Gynecol. Clin. North. Am. 2017;44(2):245-56.https://dx.doi.org/10.1016/j.ogc.2017.02.004.
- Jacobs P.A. The incidence and etiology of sex chromosome abnormalities in man. Birth Defects Orig. Artic. Ser. 1979;15(1):3-14.
- Министерство здравоохранения Российской Федерации Клинические рекомендации «Нормальная беременность». 2020. [Ministry of Health of the Russian Federation. Clinical recommendations «Normal Pregnancy». 2020. (in Russian)].
- Salomon L.J., Alfirevic Z., Audibert F., Kagan K.O., Paladini D., Yeo G., Raine-Fenning N.; ISUOG Clinical Standards Committee. ISUOG consensus statement on the impact of non-invasive prenatal testing (NIPT) on prenatal ultrasound practice. Ultrasound Obstet. Gynecol. 2014;44(1):122-3.https://dx.doi.org/10.1002/uog.13393
- Villar J., Ismail L.C., Victora C.G., Ohuma E.O., Bertino E., Altman D.G. et al. International standards for newborn weight, length, and head circumfer-ence by gestational age and sex: the newborn cross-sectional study of the INTERGROWTH-21st Project. Lancet. 2014;384(9946):857-68.https://dx.doi.org/10.1016/S0140-6736(14)60932-6.
- Beune I.M., Bloomfield F.H., Ganzevoort W., Embleton N.D., Rozance P.J., van Wassenaer-Leemhuis A.G. et al. Consensus based definition of growth restriction in the newborn. J. Pediatr. 2018;196:71-76.e1. https://dx.doi.org/10.1016/j.jpeds.2017.12.059.
- Minić S., Trpinac D., Obradović M. Incontinentia pigmenti diagnostic criteria update. Clin. Genet. 2014;85:536-42. https://dx.doi.org/10.1111/cge.12223.
- Sharma D., Shastri S., Farahbakhsh N., Sharma P. Intrauterine growth restriction - part 1. J. Matern. Fetal Neonatal Med. 2016;29(24):3977-87.https://dx.doi.org/10.3109/14767058.2016.1152249.
- Snijders R.J., Sherrod C., Gosden C.M., Nicolaides K.H. Fetal growth retardation: associated malformations and chromosomal abnormalities. Am. J. Obstet. Gynecol. 1993;168(2):547-55. https://dx.doi.org/10.1016/0002-9378(93)90491-z.
- American College of Obstetricians and Gynecologists’ Committee on Practice Bulletins—Obstetrics; Committee on Genetics; Society for Maternal-Fetal Medicine. Screening for Fetal Chromosomal Abnormalities: ACOG Practice Bulletin, Number 226. 2020;136(4):e48-e69. https://dx.doi.org/10.1097/AOG.0000000000004084.
- Сухих Г.Т., Тетруашвили Н.К., Трофимов Д.Ю., Ким Л.В., Барков И.Ю., Шубина Е.С., Парсаданян Н.Г., Федорова Н.И., Гольцов А.Ю., Александрова Н.В. Неинвазивный пренатальный ДНК-скрининг методом высокопроизводительного секвенирования у беременных с акушерской патологией. Доктор.Ру. 2017; 3(132): 11-5. [Sukhikh G.T., Tetruashvili N.K.,Trofimov D.Yu., Kim L.V., Barkov I.Yu., Shubina Ye.S., Parsadanyan N.G.,Fedorova N.I., Goltsov A.Yu., Alexandrova N.V. Next-generation sequencing technologies as a noninvasive prenatal DNA screening method in pregnant women with obstetric disorders. Doctor.Ru. 2017; 3(132): 11-5.(in Russian)].
- Linden M.G., Bender B.G., Harmon R.J., Mrazek D.A., Robinson A. 47,XXX: what is the prognosis. Pediatrics. 1988;82(4):619-30.
- Nielsen J., Wohlert M. Sex chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Birth. Defects Orig. Artic. Ser. 1990;26(4):209-23.
- MacDonald M., Hassold T., Harvey J., Wang L.H., Morton N.E., Jacobs P. The origin of 47,XXY and 47,XXX aneuploidy: heterogeneous mechanisms and role of aberrant recombination. Hum. Mol. Genet. 1994;3(8):1365-71.https://dx.doi.org/10.1093/hmg/3.8.1365.
- Drummond C.L., Gomes D.M., Senat M.V., Audibert F., Dorion A., Ville Y. Fetal karyotyping after 28 weeks of gestation for late ultrasound findings in a low risk population. Prenatal Diagnosis. 2003;23(13):1068-72.https://dx.doi.org/10.1002/pd.715.
- Robinson A., Lubs H.A., Nielsen J., Sørensen K. Summary of clinical findings: Profiles of children with 47,XXY, 47,XXX and 47,XYY karyotypes. Birth Defects Orig. Artic. Ser. 1979;15(1):261-6.
- Ardelean D., Pope E. Incontinentia pigmenti in boys: a series and review of the literature. Pediatr. Dermatol. 2006;23(6):523-7. https://dx.doi.org/10.1111/j.1525-1470.2006.00302.x.
- Fusco F., Paciolla M., Conte M.I., Pescatore A., Esposito E., Mirabelli P. et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J. Rare Dis. 2014;9:93. https://dx.doi.org/10.1186/1750-1172-9-93.
- Fusco F., Paciolla M., Pescatore A., Lioi M.B., Ayuso C., Faravelli F. et al. Microdeletion/duplication at the Xq28 IP locus causes a de novo IKBKG/NEMO/IKKgamma exon4_10 deletion in families with incontinentia pigmenti. Hum. Mutat. 2009;30(9):1284-91. https://dx.doi.org/10.1002/humu.21069.
- Fusco F., Paciolla M., Napolitano F., Pescatore A., D'Addario I., Bal E.et al. Genomic architecture at the incontinentia pigmenti locus favours de novo pathological alleles through different mechanisms. Hum. Mol. Genet. 2012;21(6):1260-71. https://dx.doi.org/10.1093/hmg/ddr556.
- Kutkowska-Kaźmierczak A., Obersztyn E., Bonnefont J.P., Rosińska-Borkowska D., Mazurczak T., Sobczyńska-Tomaszewska A., Mazurczak T. Variable clinical expression of familial incontinentia pigmenti syndrome - presentation of three cases. Med. Wieku Rozwoj. 2008;12(3):748-53.
- Berlin A.L., Paller A.S., Chan L.S. Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J. Am. Acad. Dermatol. 2002;47(2):169-87. https://dx.doi.org/10.1067/mjd.2002.125949.
- Basarab T., Dunnill M.G.S., Munn S.E., Russel-Jones R. Incontinenti pigmenti: variable disease expression within the affected family. J. Eur. Acad. Dermatol. Venerol. 1998;11(2):173-6.
- Smahi A., Courtois G., Vabres P., Yamaoka S., Heuertz S., Munnich A.et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International incontinentia pigmenti (IP) consortium. Nature. 2000.405(678):466-72.https://dx.doi.org/10.1038/35013114.
- Kawai M., Sugimoto A., Ishihara Y., Kato T., Kurahashi H. Incontinentia pigmenti inherited from a father with a low level atypical IKBKG deletion mosaicism: a case report. BMC Pediatr. 2022;22(1):378. https://dx.doi.org/10.1186/s12887-022-03444-6.
- Shibata K., Kunisada M., Miyai S., Kawamori Sh., Kurahashi H., Nishigori C. Incontinentia pigmenti in a female infant with somatic mosaicism due to the IKBKG variant. J. Dermatol. 2021;48(12):e577-e578.https://dx.doi.org/10.1111/1346-8138.16141.
- Fusco F., Conte M.I., Diociauti A., Bigoni S., Branda M.F., Ferlini A. et al. Unusual father-to-daughter transmission of incontinentia pigmenti due to mosaicism in IP males. Pediatrics. 2017;140(3):e20162950.https://dx.doi.org/10.1542/peds.2016-2950.
- Poziomczyk C.S., Recuero J.K., Bringhenti L., Maria F.D., Campos C.W., Travi G.M. et al. Incontinentia pigmenti. An. Bras. Dermatol. 2014;89(1):26-36. https://dx.doi.org/10.1590/abd1806-4841.20142584.
- Stevenson R.E., Hall J.G., ed. Human malformations and related anomalies. Oxford monographs on medical genetics -- no. 52. 2nd ed. Oxford University Press; 2006. 1495 p.
- Пушкарева Ю.Э., Федоров И.А. Случай синдрома Блоха-Сульцбергера у новорожденного ребенка. Вестник Совета молодых ученых и специалистов. 2017;3(18):57-60. [Pushkareva Yu.E., Fedorov I.A. The case of the Bloch-Sutzberger syndrome in a newborn child. Bulletin of the Council of young scientists and specialists. 2017;3(18):57-60. (in Russian)].
- Земсков М.А., Котлова В.Б., Долгих В.С., Иванова С.Н., Ромашова В.В. Синдром Блоха-Сульцбергера (недержание пигмента): редкий генодерматоз с поражением глаз. Consilium Medicum. 2019;21(12.2):52-3. [Zemskov M.A., Kotlova V.B., Dolgikh V.S. et al. Bloch-Sulzberger syndrome (incontinentia pigmenti) – a rare genodermatosis with injury to eyes. Consilium Medicum. 2019;21(12.2):52-3. (in Russian)]. https://dx.doi.org/10.26442/24143537.2019.2.190363.
- Черникова Т.И., Шепилов Л.А., Васина Т.Н., Зубцова Т.И., Ставцева С.Н., Вислобоков А.В. Тяжелая форма синдрома Блоха-Сульцбергера у новорожденного ребенка. Российский вестник перинатологии и педиатрии. 2014;59(5):59-62. [Chernikova T.I., Shepilov L.A., Vasina T.N., Zubtsova T.I., Stavtseva S.N., Vislobokov A.V. Severe Bloch-Sulzberger syndrome in a newborn baby. Russian Bulletin of Perinatology and Pediatrics. 2014;59(5):59-62. (in Russian)].
- Артёмчик Т.А., Музыченко А.П., Устинович А.А., Крастелёва И.М., Ляшевич Е.В., Кастусик С.В. Синдром Блоха-Сульцбергера. Клинический случай. Дерматовенерология. Косметология. 2018;4(3):347-52. [Artemchik T., Muzychenko A., Ustinovich A., Krasteljova I., Lyashevich E., Kastushik S. Bloch-Sulzberger syndrome. Clinical case. Dermatovenerology. Cosmetology. 2018;4(3):347-52. (in Russian)].
- Краснова Н.В., Чернова Т.А., Алексеева И.В., Гималиева Г.Г., Синицына Л.Г., Мисякова Т.Ю. Клинический случай синдрома Блоха-Сульцбергера. Вестник дерматологии и венерологии. 2020;96(3):63-7. [Krasnova N.V., Chernova T.A., Alekseeva I.V., Gimalieva G.G., Sinitsyna L.G.,Misyakova T.Yu. Clinical case of Bloch-Sulzberger syndrome. Bulletin of dermatology and venereology. 2020;96(3):63-7. (in Russian)].https://dx.doi.org/10.25208/vdv1117.
- Иванова И.Е., Ногтева Л.Г., Горячкина Л.А., Алексеева И.В., Абрукова А.В., Гималиева Г.Г. Синдром Блоха-Сульцбергера у детей. Здравоохранение Чувашии. 2020;(3):32-40. [Ivanova I.E., Nogteva L.G., Goryachkina L.A., Alekseeva I.V., Abrukova A.V., Gimalieva G.G. Bloch-Sulzberger syndrome in children. Healthcare in Chuvashia. 2020;(3):32-40. (in Russian)].
- Yuan F., Zhu F.N., Liu X.J., Li J., Xu H.T. Incontinentia pigmenti: a case report of early clinical symptoms in a lack of family inheritance positive result. Clin. Cosmet. Investig. Dermatol. 2023;16:1209-14. https://dx.doi.org/10.2147/CCID.S407506.
- Nirmalasari D.A., Tabri F., Waspodo N., Rimayani S., Adriani A. Incontinentia pigmenti / Bloch-Sulzberger syndrome: a case report. Acta Dermatovenerol Alp. Pannonica Adriat. 2022;31(1):39-41.
- Михайлин Е.С., Иванова Л.А., Савицкий А.Г., Королева Л.И., Касьянова Д.С. Случай беременности и родов у пациентки с синдромом Блоха-Сульцбергера. Российский вестник акушера-гинеколога. 2017;17(2):47-9. [Mikhailin E.S., Ivanova L.A., Savitsky A.G., Koroleva L.I., Kasyanova D.S. A case of pregnancy and delivery in a patient with Bloch-Sulzberger syndrome. Russian Bulletin of Obstetrician-Gynecologist. 2017;17(2)47-9. (in Russian)]. https://dx.doi.org/10.17116/rosakush201717247-49.
- Kim M.J., Lyu S.W., Seok H.H., Park J.E., Shim S.H., Yoon T.K. A healthy delivery of twins by assisted reproduction followed by preimplantation genetic screening in a woman with X-linked dominant incontinentia pigmenti. Clin. Exp. Reprod. Med. 2014;41(4):168-73. https://dx.doi.org/10.5653/cerm.2014.41.4.168.
- Pettigrew R., Kuo H.C., Scriven P., Rowell P., Pal K., Handyside A. et al. A pregnancy following PGD for X-linked autosomal dominant incontinentia pigmenti (Bloch-Sulzberger syndrome): case report. Human Reproduction. 2000;15(12):2650-2. https://dx.doi.org/10.1093/humrep/15.12.2650.
- Бахлыкова Е.А., Комсюкова К.Ф., Матусевич С.Л., Жвавый П.Н., Ковкова Г.Ю., Немцова И.В. Случай сочетания синдрома Блоха-Сульцбергера с синдромом Дауна. Клиническая дерматология и венерология. 2018;17(4):30-4. [Bakhlykova E.A., Komsukova K.F., Matusevich S.L., Zhvavy P.N., Kovkova G.Yu., Nemtsova I.V. The case of concomitant Bloch—Sulzberger and Down syndromes. Klinicheskaya Dermatologiya i Venerologiya. Russian journal of clinical dermatology and venerology. 2018;17(4):30-4.(in Russian)]. https://dx.doi.org/10.17116/klinderma20181704130.
- Parrish J.E., Scheuerle A.E., Lewis R.A., Levy M.L., Nelson D.L. Selection against mutant alleles in blood leukocytes is a consistent feature in incontinentia pigmenti type 2. Hum. Mol. Genet. 1996;5(11):1777-83.https://dx.doi.org/10.1093/hmg/5.11.1777.
- Sugawara N., Maeda M., Manome T., Nagai R., Araki Y. Patients with 47, XXX karyotype who experienced premature ovarian failure (POF): two case reports. Reprod. Med. Biol. 2013;12(4):193-5. https://dx.doi.org/10.1007/s12522-013-0158-9.
- Skordis N., Ferrari E., Antoniadou A., Phylactou L.A., Fanis P., Neocleous V. GnRH-dependent precocious puberty manifested at the age of 14 months in a girl with 47, XXX karyotype. Hormones (Athens). 2017;16(3):318-21.https://dx.doi.org/10.14310/horm.2002.1740.
- Wade B.S., Joshi S.H., Reuter M., Blumenthal J.D., Toga A.W., Thompson P.M., Giedd J.N. Effects of sex chromosome dosage on corpus callosum morphology in supernumerary sex chromosome aneuploidies. Biol. Sex Differ. 2014;5:16. https://dx.doi.org/10.1186/s13293-014-0016-4.
- Otter M., Schrander-Stumpel C.T., Didden R., Curfs L.M.G. The psychiatric phenotype in triple X syndrome: new hypotheses illustrated in two cases. Dev. Neurorehabil. 2012;15(3):233-8. https://dx.doi.org/10.3109/17518423.2012.655799.
- Tartaglia N.R., Howell S., Sutherland A., Wilson R., Wilson L. A review of trisomy X (47,XXX). Orphanet J Rare Dis. 2010;5:8. https://dx.doi.org/10.1186/1750-1172-5-8.
- Jańczewska I., Wierzba J., Jańczewska A., Szczurek-Gierczak M., Domżalska-Popadiuk I. Prematurity and low birth weight and their impact on childhood growth patterns and the risk of long-term cardiovascular sequelae. Children (Basel). 2023;10(10):1599. https://dx.doi.org/10.3390/children10101599.
- Halevy J., Peretz R., Ziv-Baran T., Katorza E. Fetal brain volumes and neurodevelopmental outcome of intrauterine growth restricted fetuses. Eur. J. Radiol. 2023;168:111143. https://dx.doi.org/10.1016/j.ejrad.2023.111143.
- Korzeniewski S.J., Sutton E., Escudero C., Roberts J.M. The Global Pregnancy Collaboration (CoLab) symposium on short- and long-term outcomes in offspring whose mothers had preeclampsia: A scoping review of clinical evidence. Front. Med. (Lausanne). 2022;9:984291. https://dx.doi.org/10.3389/fmed.2022.984291.
- Rodrigues M., Pandya A.G. Hypermelanoses. In: Kang S., Amagai M., Bruckner A.L., Enk A.H., Margolis D.J., McMichael A.J. et al., eds. Fitzpatrick’s dermatology. 9th ed. New York: McGraw-Hill; 2019. 1368 p.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J. Pediatr. Health Care. 2017;31(6):e45-e52.https://dx.doi.org/10.1016/j.pedhc.2017.07.003.
- Bodemer C., Diociaiuti A., Hadj-Rabia S., Robert M.P., Desguerre I., Manière M.C. et al. Multidisciplinary consensus recommendations from a European network for the diagnosis and practical management of patients with incontinentia pigmenti. J. Eur. Acad. Dermatol. Venerol. 2020;34(7):1415-24.https://dx.doi.org/10.1111/jdv.16403.
Received 12.12.2023
Accepted 27.12.2023
About the Authors
Anna S. Bolshakova, Geneticist at the Department of Clinical Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-24-11, a_bolshakova@oparina4.ru, 117997, Russia, Moscow, Ac. Oparin str., 4, https://orcid.org/0000-0002-7508-0899Ilya Yu. Barkov, PhD, Head of the Laboratory of Prenatal DNA Screening, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-24-10, i_barkov@oparina4.ru, 117997, Russia, Moscow, Ac. Oparin str., 4.
Natalia A. Frankevich, PhD, Senior Researcher, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology,
Ministry of Health of Russia, natasha-lomova@yandex.ru, 117997, Russia, Moscow, Ac. Oparin str., 4, https://orcid.org/0000-0002-6090-586X
Tamara A. Yarygina, PhD, Diagnostic Medical Sonographer, Researcher at the Perinatal Cardiology Center, A.N. Bakulev National Medical Research Center of Cardiovascular Surgery, Ministry of Health of Russia, 121552, Russia, Moscow, Roublyevskoe Shosse, 135, +7(495)414-78-75; Associate Professor at the Department of Ultrasound Imaging of the Faculty of Continuing Medical Education of the Medical Institute, Patrice Lumumba Peoples' Friendship University of Russia, 127015, Russia, Moscow, Pistsovaya str., 10, tamarayarygina@gmail.com, https://orcid.org/0000-0001-6140-1930
Roman G. Shmakov, Dr. Med. Sci., Professor of the Russian Academy of Sciences, Director of the Institute of Obstetrics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-72-00, r_shmakov@oparina4.ru, 117997, Russia, Moscow, Ac. Oparin str., 4.
Corresponding author: Tamara A. Yarygina, tayarygina@bakulev.ru
Similar Articles

