
Скелетные дисплазии – это нарушения развития костной ткани, представляющие собой гетерогенную и сложную группу нарушений скелетогенеза. Современная классификация скелетных дисплазий выделяет 461 заболевание, суммарная частота которых составляет более чем 1:5000 [1, 2].
Танатофорная дисплазия (ТД) – это аутосомно-доминантное врожденное заболевание, связанное с первичной дисплазией костей, характеризующееся микромелией, макроцефалией и платиспондилией. Для данного заболевания характерны узкая грудная клетка, короткие ребра, недоразвитые легкие и увеличенная голова с большим лбом и выдающимися широко расставленными глазами [3, 4]. Частота ТД среди живорожденных в общей популяции составляет 1:33 000–1:47 000 [5]. Частота пренатального выделения выше и может достигать 1:12 000 [5]. Эпидемиологических данных по частоте встречаемости ТД в России в научной литературе не представлено. Большинство младенцев с ТД рождаются мертворожденными или умирают вскоре после рождения от дыхательной недостаточности [3], хотя есть сообщения о людях, проживших более 16 лет при существенной медицинской поддержке (все они являются тяжелыми инвалидами) [6, 7]. Описаны две основные формы ТД: ТД I и II типа. ТД I типа отличается наличием искривленных бедренных костей и уплощенных костей позвоночника (платиспондилия). ТД II типа характеризуется прямыми бедренными костями и черепом в форме клеверного листа [8, 9].
Основной причиной ТД на сегодняшний день считаются патогенные варианты в гене рецептора фактора роста фибробластов FGFR3. Это заболевание с аутосомно-доминантным типом наследования, практически все описанные случаи являются спорадическими [2]. Ген FGFR3 картирован на хромосоме 4p16.3 и состоит из 18 кодирующих экзонов. Белок, кодируемый FGFR3, является трансмембранным рецептором тирозинкиназы, связывающим факторы роста фибробластов (fibroblast growth factor receptor-3, OMIM#134934) [2, 10]. Патогенные варианты в гене FGFR3 приводят к сверхактивации рецептора, что ведет к ингибированию пролиферации хондроцитов и нарушению эндохондриальной оссификации [11] и может быть ассоциировано с ахондроплазией (Achondroplasia, OMIM #100800), гипохондроплазией (Hypochondroplasia, OMIM #146000,), ТД I и II типов (Thanatophoric dysplasia type I, OMIM #187600, Thanatophoric dysplasia type II, OMIM #187601), синдромом Крузона с черным акантозом (Crouzon syndrome with acanthosis nigricans, OMIM #612247), синдромом Мюнке (Muenke syndrome, OMIM #602849) и др.
Частота ТД составляет 0,2–0,5:10 000 живорождений [2]. Описано по крайней мере 11 различных патогенных вариантов FGFR3, которые были идентифицированы при TД I типа. Согласно общепопуляционным данным, наиболее частыми патогенными генетическими вариантами, ответственными за развитие ТД I, типа являются p.Arg248Сys и p.Tyr373Cys (NM_142000.5); эти патогенные варианты вместе составляют около 90% выявленных случаев [10]. У пациентов с TД II типа выявляется преимущественно аминокислотная замена FGFR3 p.Lys650Glu (К650Е), NM_142000.5, но в последние годы было описано несколько новых генетических вариантов [11, 12].
Цель исследования: обосновать необходимость проведения молекулярно-генетической пренатальной диагностики ТД у женщин с ультразвуковыми маркерами скелетных аномалий у плода с помощью описания клинических наблюдений.
Материалы и методы
Для описания клинических наблюдений использовалась медицинская документация двух женщин. Обе пациентки дали согласие на использование данных результатов обследования для научных исследований и публикации в научных изданиях в деперсонифицированном виде. Ретроспективное исследование одобрено на заседании этического комитета ГБУЗ СО «Екатеринбургский клинический перинатальный центр» (протокол № 2 от 26.05.2023).
Пациентки были направлены в ГАУЗ СО «Клинико-диагностический центр “Охрана здоровья матери и ребенка”» (КДЦ «ОЗМР»), г. Екатеринбург, в 2019–2020 гг. для проведения экспертного ультразвукового исследования (УЗИ) в комплексе пренатальной диагностики I триместра, при котором была обнаружена патология развития плода. Обеим женщинам было рекомендовано проведение инвазивной диагностики (аспирация ворсин хориона). Первоначально было проведено цитогенетическое исследование. Далее образцы ворсин хориона от плодов данных пациенток после инвазивной процедуры были доставлены в лабораторию молекулярной диагностики ГБУЗ СО «КДЦ «ОЗМР» и заморожены.
Методом прямого секвенирования по Сэнгеру ретроспективно осуществлен поиск патогенных вариантов в кодирующей области гена FGFR3. Геномную ДНК выделяли из размороженных пренатальных образцов ворсин хориона на автоматизированной станции MagNa Pure LC 2.0 (Roche, США) с использованием коммерческого набора MagNa Pure LC DNA Isolation Kit I (Roche, США) согласно протоколу производителя. Кодирующие участки гена FGFR3 амплифицировали с помощью полимеразной цепной реакции (ПЦР). Амплификацию проводили на программируемом термоциклере MyCycler (BioRad, США) с использованием TaqДНК-полимеразы («СибЭнзим») и праймеров, описанных Xue Y. et al. (2014) [8]. Смесь для ПЦР (25 мкл) содержала 1х реакционный буфер (60 mM Tris-HCl, 1,5 mM MgCl2, 25mM KCl, 10mM 2-меркаптоэтанол, 0,1% Тритон Х-100), 10 пМ каждого олигопраймера, 400 мкМ каждого дезоксинуклеотида, 1,25 единицы термофильной ДНК-полимеразы и 0,2–1 мкг геномной ДНК. ПЦР проводили в следующем режиме: первоначальная денатурация при 95°С – 5 минут, далее 30 циклов: 95°С – 30 с, 71°С – 30 с, 72°С – 45 с, заключительная элонгация при 72°С в течение 5 мин. Детекцию ПЦР-продуктов проводили в 8% полиакриламидном геле (соотношение акриламида/бисакриламида 39:1). Секвенирование ПЦР-продуктов, полученных в ходе амплификации, проводили методом секвенирования ДНК по Сэнгеру (по обеим цепям) на секвенаторе ABI 3500 DNA Analyzer (Applied Biosystems, США) с оригинальными реагентами, согласно рекомендациям производителя. Анализ результатов секвенирования осуществлялся с помощью программ VariantReporter и SeqA6 (https://www.thermofisher.com).
Для проведения клинического секвенирования экзома материал был направлен в ООО «Геномед». Анализ ДНК проводился по технологии секвенирования нового поколения методом парно-концевого прочтения. Для пробоподготовки использовалась методика селективного захвата участков ДНК, относящихся к кодирующим областям генов с известным клиническим значением и описанных в данных ОMIM. Среднее покрытие целевых участков секвенирования в исследуемых генах составляло не менее 70х.
Описание
Клиническое наблюдение № 1
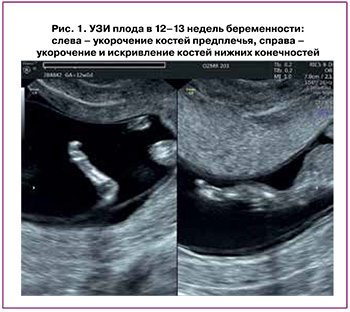
Пациентка П., 22 года, обратилась в КДЦ «ОЗМР» в сроке беременности 12–13 недель для проведения комплекса пренатальной диагностики I триместра. При экспертном УЗИ (рис. 1) были отмечены отсутствие визуализации носовой кости, укорочение костей конечностей.
Учитывая наличие маркеров хромосомных аномалий по данным УЗИ, беременная была направлена на инвазивную пренатальную диагностику – проведена аспирация ворсин хориона. В результате было определено, что кариотип клеток хориона – 46, ХY (диплоидный мужской кариотип), данных за наличие хромосомной аномалии не получено.
В гестационном сроке 19–20 недель при проведении УЗИ выявлены множественные врожденные пороки развития (ВПР) у плода: ВПР центральной нервной системы – межполушарный вариант голопрозэнцефалии (ширина бокового желудочка мозга составила 18,8 мм, размер третьего желудочка – 8,2 мм, желудочки сливались между собой, образуя общую полость (рис. 2)), врожденный порок сердца – дефект межжелудочковой перегородки, укорочение и искривление костей конечностей (из-за чего возникло подозрение на наличие ТД).

Учитывая тяжесть выявленных ВПР, беременная была направлена на перинатальный консилиум, где ей было разъяснено, что имеются медицинские показания со стороны плода для прерывания беременности. Однако консультируемая от прерывания беременности отказалась.
Беременность протекала с осложнениями. При беременности развился холестатический гепатоз беременных (с 30 недель), диагностирована умеренная преэклампсия (в 35–36 недель). У беременной была резус-отрицательная кровь, в 28 недель проведена антенатальная иммунопрофилактика.
В 36–37 недель гестации произошли преждевременные оперативные роды, родился мальчик весом 3500 г, длиной 40 см, с оценкой по шкале Апгар 2/5 баллов с множественными ВПР: ВПР центральной нервной системы (межполушарный вариант голопрозэнцефалии), врожденный порок сердца – дефект межжелудочковой перегородки, кардиомегалия, ВПР опорно-двигательного аппарата: укорочение костей верхних и нижних конечностей, гипоплазия грудной клетки, вторичная гипоплазия легких. Окружность головы ребенка при рождении составила 45 см, окружность груди – 30 см. При проведении рентгенографии были выявлены гипоплазия легких, аномалии развития ребер, позвоночника, трубчатых костей конечностей и костей таза. Ребенок был переведен в отделение реанимации и интенсивной терапии, на 5-е сутки его жизни мать отказалась от дальнейшего пребывания в стационаре; в связи с отказом от дальнейшей госпитализации была выписана домой. Ребенок умер дома в неонатальном периоде. Диагноз «танатофорная дисплазия» был выставлен только после родов.
При последующем исследовании ДНК, выделенной из ворсин хориона, полученных в результате инвазивной процедуры в 12–13 недель беременности у пациентки П., в 14-м экзоне гена FGFR3 выявлен патогенный вариант c.1948 A>G (p.Lys650Glu) в гетерозиготном состоянии (рис. 3). Данный вариант p.K650E находится в пределах критической области активации тирозинкиназы, аминокислотная замена в этом кодоне приводит к сильному аутофосфорилированию нескольких тирозиновых остатков и увеличению сигнальной способности тирозинкиназы, вызывая нарушения роста костей, в том числе за счет преждевременного окостенения. Данный вариант был описан ранее у пациентов с TД II типа как патогенный [11, 13, 14].

Клиническое наблюдение № 2
Пациентка С., 21 год, обратилась в сроке беременности 14–15 недель. В комплексе пренатальной диагностики I триместра при проведении УЗИ выявлены аномалии опорно-двигательного аппарата у плода: трубчатые кости рук и ног укорочены и деформированы, грудная клетка гипоплазирована (рис. 4), форма черепа – «клеверообразная», что характерно для ТД.
Предварительный диагноз – ТД, учитывая особенности формирования опорно-двигательного аппарата у плода. Так же, как и в первом случае, с целью определения прогноза для потомства проведена аспирация ворсин хориона для кариотипирования и молекулярно-генетического исследования материала плода; кариотип клеток хориона 46,ХY. Беременная была направлена на пренатальный консилиум, было дано заключение о наличии медицинских показаний для прерывания беременности. С согласия пациентки беременность была прервана в сроке 14–15 недель.
Впоследствии ДНК, выделенная из ворсин хориона, была направлена на дополнительное исследование. В результате молекулярно-генетической диагностики методом полного секвенирования экзома в гене FGFR3 в гетерозиготном состоянии обнаружена однонуклеотидная замена с.742 С>T (p.Arg248Сys), ранее описанная как патогенная [15]. Наличие данного патогенного варианта у плода было подтверждено лабораторией молекулярной диагностики ГАУЗ СО «КДЦ “Охрана здоровья матери и ребенка”» методом прямого секвенирования по Сэнгеру 7-го экзона гена FGFR3 (рис. 5).
Семье было разъяснено, что риск ТД для потомства в дальнейшем незначительно превышает общепопуляционный. Через 1,5 года после прерывания беременности женщина забеременела повторно. В результате родился живой доношенный ребенок без пороков развития.
В обоих представленных клинических наблюдениях не удалось провести обследование членов семьи для установления происхождения выявленных патогенных вариантов; однако, учитывая аутосомно-доминантный тип наследования указанных нозологий, мы склонны отнести данные варианты к статусу de novo. В научной литературе выявленные нами генетические варианты описаны как патогенные, поэтому обследование родителей не является обязательным.
Обсуждение
Пренатальная сонографическая диагностика скелетных дисплазий плода представляет собой сложную задачу из-за их редкости, множества дифференциальных диагнозов с перекрывающимися признаками и фенотипической вариабельности [9]. ТД является наиболее распространенной скелетной дисплазией с летальным исходом в неонатальном периоде. Точный диагноз ТД может быть установлен только с помощью молекулярно-генетических методов ДНК-диагностики единственного известного гена FGFR3, ответственного за развитие данной нозологии [5, 10, 14]. Молекулярная диагностика в семьях, столкнувшихся с диагнозом ТД необходима как для планирования последующего деторождения, так и для подтверждения диагноза при текущей беременности. Например, в недавней работе Audu L. et al. (2022) описано 2 случая, когда ТД у плода была пропущена, хотя имелся ряд ультразвуковых признаков патологии скелета; в результате родились дети с характерным внешним видом, которые в течение 36 ч умерли из-за того, что у них развился респираторный дистресс-синдром [16]. Не секрет, что будущие родители могут испытывать сомнения в правильности диагноза ТД у плода лишь на основании данных УЗИ-диагностики, без молекулярно-генетического подтверждения, и отказаться от раннего прерывания беременности, приняв решение о дальнейшем вынашивании беременности в надежде, что диагноз не подтвердится. Повышенная тревожность женщины в течение пролонгированной беременности, а затем и неминуемые внутриутробная или неонатальная потеря ребенка подвергают семью сильнейшему психологическому стрессу. Кроме психологических, возможны и физические последствия вынашивания беременности с неблагоприятным перинатальным исходом. В описываемом нами клиническом наблюдении № 1 беременная отказалась от прерывания беременности; в результате подвергала себя рискам, связанным с осложненным течением беременности. Были проведены оперативные роды, следовательно, пациентка имеет рубец на матке после операции кесарева сечения, что повышает риск для ее здоровья при последующих беременностях. ДНК-диагностика, выполненная на ранних сроках беременности, могла бы своевременно подтвердить неблагоприятный прогноз для плода и наличие медицинских показаний для прерывания беременности.
После прерывания беременности или рождения ребенка с признаками ТД важным вопросом, который волнует родителей, является вероятность повторения подобной ситуации в дальнейшем. Неопределенный прогноз способствует появлению опасений у супругов вновь получить тяжелую психологическую травму, связанную с потерей ребенка, и в результате возможен отказ супругов от деторождения. При подтверждении наличия патогенных вариантов в гене FGFR3 у плода родителям разъясняется, что данные изменения в генотипе, как правило, появляются de novo, то есть не являются унаследованными от них самих. В ряде случаев, если супруги настаивают, для их психологического комфорта может быть проведен поиск патогенных вариантов FGFR3 у них самих (для подтверждения de novo статуса обнаруженного патогенного варианта у плода).
В то же время имеется ряд заболеваний, имитирующих ТД, но имеющих иной тип наследования. В случае, если подтверждено наличие у плода патогенного варианта в гетерозиготном состоянии в гене FGFR3, риск скелетной дисплазии для потомства в будущем в обследуемой семье незначительно превышает общепопуляционный. Возможно наличие гонадного мозаицизма у одного из супругов, однако для ТД описаны лишь единичные случаи такой особенности [3, 17], и даже с учетом вероятности гонадного мозаицизма риск ТД для сибсов составляет не более 0,02% [18]. Тем не менее необходимо проводить дифференциальную диагностику с наследственной патологией скелета с аутосомно-рецессивным типом наследования. Например, к подобным ТД заболеваниям могут приводить гомозиготные и компаунд-гетерозиготные патогенные варианты в гене RMRP [8]. Описано и несколько видов несовершенного остеогенеза с аутосомно-рецессивным типом наследования, например, ассоциированный с патогенными вариантами гена PLOD2 [19]. В этом случае риск для сибсов пробанда резко возрастает и составит 25%.
В ряде работ при выявлении признаков скелетной дисплазии у плода предлагается проведение клинического секвенирования экзома [20]. Однако мы полагаем, что в качестве теста первой линии при наличии признаков ТД следует предлагать таргетный поиск патогенных вариантов в гене FGFR3, так как ТД – это наиболее распространенная скелетная дисплазия с летальным исходом [3]. В нашей работе в обоих случаях были выявлены известные патогенные варианты в указанном гене. В исследовании Wang L. et al. (2022) обсуждалась целесообразность проведения клинического секвенирования экзома при выявлении задержки роста плода и/или укорочении трубчатых костей; однако авторы пришли к выводу, что на сегодняшний день клиническая полезность подобной тактики сомнительна [20]. В то же время, учитывая вероятность фенокопий, мы полагаем, что требуются дальнейшие исследования для того, чтобы окончательно поставить точку в этом вопросе.
Заключение
Подтверждение диагноза «танатофорная дисплазия» с помощью молекулярно-генетических методов исследования при наличии ультразвуковых признаков патологии скелета плода необходимо для уточнения наличия медицинских показаний для прерывания беременности и для определения прогноза для будущего потомства в семье.


