

Uterus didelphys, longitudinal vaginal septum and diminished ovarian reserve in a patient with MYRF-associated disease
Tsabai P.N., Batyrova Z.K., Kumykova Z.Kh., Kovalskaya V.A., Sidorchuk M.A., Goltsov A.Yu., Sadelov I.O., Tolmacheva E.R., Uvarova E.V., Sukhikh G.T., Trofimov D.Yu.
Background: Congenital abnormalities of female reproductive organs result from the abnormal formation, fusion or resorption of Müllerian ducts during the intrauterine period and can lead to adverse reproductive outcomes. A number of studies have identified a higher incidence of ovarian dysfunction in patients with uterine abnormalities; however, other studies do not support this correlation. Uterine defects are frequently associated with syndromic pathologies; yet the etiology of the most prevalent Müllerian anomalies remains unclear or multifactorial.
Case report: This paper presents a clinical observation of a patient with uterus didelphys, longitudinal vaginal septum and diminished ovarian reserve as part of cardiac-urogenital syndrome caused by a likely pathogenic variant p.Ala440ThrfsTer2 in the MYRF gene. The patient was recommended to consider assisted reproductive technologies with preimplantation genetic testing to prevent the birth of an affected child.
Conclusion: This case illustrates the pleiotropic effect of the MYRF transcription factor. Pathogenic variants in the MYRF gene can impair the development of both Müllerian derivatives and ovaries, and lead to diminished ovarian reserve, which is also accompanied by dysfunction of other organs and systems. MYRF should be considered as a candidate gene for a variety of uterine abnormalities, diminished ovarian reserve and premature ovarian insufficiency in female patients in the presence or absence of extragenital manifestations.
Authors’ contributions: Sukhikh G.T., Uvarova E.V., Trofimov D.Yu. – developing the concept and design of the study; Tsabai P.N., Batyrova Z.K., Kumykova Z.Kh., Kovalskaya V.A. – consulting patients, collecting and processing material; Goltsov A.Yu. – performing laboratory studies; Sadelov I.O., Tolmacheva E.R. – conducting bioinformatics analysis; Tsabai P.N., Sidorchuk M.A., Tolmacheva E.R. – writing the text; Batyrova Z.K., Kumykova Z.Kh. – editing the article.
Conflicts of interest: The authors declare that there are no conflicts of interest.
Funding: This study was supported by a grant from the Russian Science Foundation [Grant No. 24-14-00460].
Ethical Approval: The study was approved by the Ethical Review Board of the Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia.
Patient Consent for Publication: The patient and her parents provided the written informed consent that all information (including photographs) could be used for research purposes.
For citation: Tsabai P.N., Batyrova Z.K., Kumykova Z.Kh., Kovalskaya V.A., Sidorchuk M.A., Goltsov A.Yu., Sadelov I.O., Tolmacheva E.R., Uvarova E.V., Sukhikh G.T., Trofimov D.Yu. Uterus didelphys, longitudinal vaginal septum and diminished ovarian reserve in a patient with MYRF-associated disease.
Akusherstvo i Ginekologiya/ bstetrics and Gynecology. 2025; (4): 171-177 (in Russian)
https://dx.doi.org/10.18565/aig.2024.321
Keywords
case report
cardiac-urogenital syndrome
MYRF
uterus didelphys
diminished ovarian reserve
vaginal septum
Müllerian anomaly
whole exome sequencing
Congenital abnormalities of female reproductive structures result from the abnormal formation, fusion or resorption of Müllerian ducts during fetal life and can lead to adverse reproductive outcomes. Müllerian ducts are presented by paired tubular structures that undergo a complex process of differentiation to give rise to the fallopian tubes, uterus, cervix, and upper two-thirds of vagina. Anomalies of the müllerian ducts include hypoplasia/agenesis of uterus (müllerian aplasia, or Mayer-Rokitansky-Kuster-Hauser syndrome) and various forms of incomplete müllerian fusion: unicornuate uterus, uterus didelphys, bicornuate uterus, septate uterus, and arcuate uterus. Uterine anomalies affect approximately 5.5% of the female population. Their prevalence increases in women with infertility (8%) or a history of pregnancy loss (13.3%) with rates as high as 24.5% in those experiencing both issues [1].
Uterine anomalies interfere with normal implantation and placenta formation, cause reduced conception rate, miscarriage, preterm labor or fetal malpresentation. Some studies indicate that ovarian dysfunction is more prevalent in women with uterine anomalies, especially Mayer–Rokitansky–Kuster–Hauser syndrome, although this evidence is controversial [2, 3]. Ovarian malposition is frequently seen in patients with uterine anomalies, although this does not seem to contribute to their dysfunction [4, 5]. The interconnection between uterus and ovaries embryogenesis is supported by the fact that approximately 17% of patients with unilateral ovarian agenesis have concomitant uterine abnormalities, while the etiology of ovarian absence is indeterminate in 27% or embryologic in 21% [6]. Very little is known about common pathways in gonadal and genital organs development.
Uterine anomalies are often associated with extragenital pathology [7]. Some of these associations belong to defined genetic syndromes, while etiology of most mullerian anomalies is deemed mainly multifactorial. However, there are reports of uterine defects running in families, suggesting that specific genetic mutations may cause these defects, and several candidate genes are reported so far [8]. The genetic causes of ovarian dysgenesis, diminished ovarian reserve (DOR) and premature ovarian insufficiency (POI) are also under investigation, and many syndromes are associated with ovarian dysfunction [9]. The development of indifferent gonads and Müllerian ducts are different biological processes, and the coincidence of uterine and ovarian anomalies is frequently due to primary gonadal failure that leads to mullerian regression or hypoplasia [10]. However, the same genes may be implicated in both uterine and ovarian development (e.g., EMX2 [11], TP63 [12], FOXL2 [13]).
Here, we report a female patient who had apparently unrelated duplication of uterus and small ovaries, which turned out to be cardiac-urogenital syndrome caused by a likely pathogenic variant p.Ala440ThrfsTer2 in the MYRF gene.
Case report
The institutional Ethical review board at Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology approved this study. This case study was carried out according to the Code of Ethics of the World Medical Association (Declaration of Helsinki). The patient and her parents gave written informed consent for the use of any data for scientific purposes.
A 16-years-old girl first referred to the adolescent gynecology department of the Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology with complaints of irregular menstrual cycles (from 16 to 39 days) since menarche at 12 years old. Her medical history was remarkable for preterm birth at 35-36 weeks of gestation via C-section, butterfly vertebrae Th10 with kyphosis and C-shaped right scoliosis (spinal deformity surgically corrected twice at the age of 13 and 14), and congenital bilateral high hypermetropia (+10 D). She reached developmental milestones in time. At the age of 15 she was diagnosed with uterus didelphys.
On the examination she was 163-cm tall female with normally developed secondary female sex characteristics (Tanner stage 5 breast development and pubic hair), normal external genitalia and two palpable uteri on digital rectal exam. At pelvic ultrasound (US) a double uterus and cervix were found, bilateral ovaries had typical localization, but appeared small with diminished antral follicular count (left ovary volume 2.4 ml, right ovary volume 2.6 ml). MRI of pelvic organs was not performed as spine constructions were incompatible with MRI. During vaginoscopy a vaginal septum up to 3 mm thick was visualized at a distance of 1.5 cm from the hymen. The septum was flanked by conical cervices with ectopy, and scanty blood was discharging from cervical canals. Abdominal, renal and heart ultrasound were normal. Laboratory tests showed normal levels of follicle-stimulating hormone (FSH) – 8.7 IU/l, luteinizing hormone – 3.5 IU/l, estradiol – 118 pmol/l, testosterone – 1.47 nmol/l, dehydroepiandrosterone sulfate – 9.3 µmol/l, and low anti-Müllerian hormone (AMH) level – 0.46 ng/ml, which was consistent with diminished ovarian reserve seen on US. Thyroid stimulating hormone (2.42 mIU/l) and prolactin levels were normal. Screening for anti-ovarian autoantibodies, 21-hydroxylase autoantibodies and thyroid peroxidase antibodies was negative.
In a course of one-and-a-half-year laboratory findings remained stable, with FSH 6.7 IU/l, 6.5 IU/l, 6.6 IU/l, and AMH 0.63 ng/ml, 0.4 ng/ml, 0.52 ng/ml, respectively. At the age of 17 the dissection of the vaginal septum was performed to form a single vaginal cavity. At the age of 18, the patient referred to a reproductive specialist in our center to discuss possible assisted reproductive technologies (ART) options for oocyte cryopreservation in future.
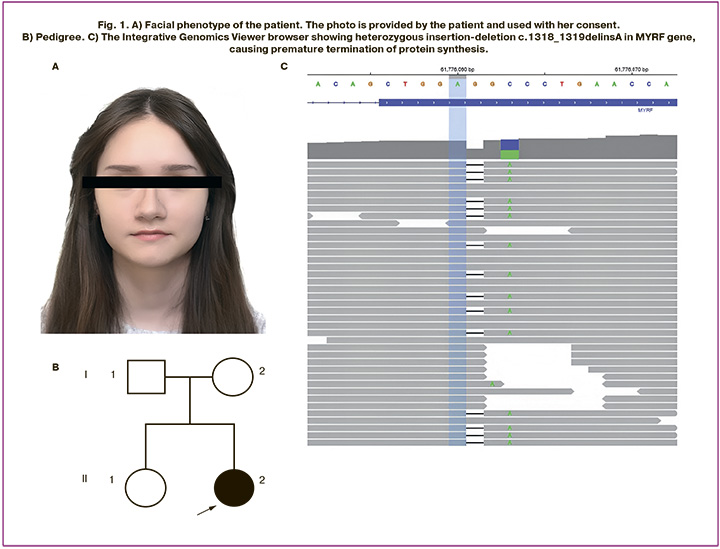
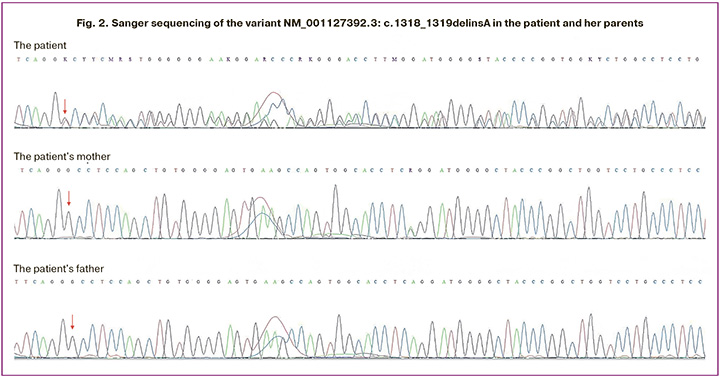
At genetic examination the patient appeared not to have dysmorphic features (Fig. 1A). Family history was unremarkable (Fig. 1B). The genetic testing was ordered to investigate for causes of congenital uterine anomaly and diminished ovarian reserve. Karyotype was normal female 46,XX, the number of CGG repeats in the FMR1 gene was 24 on one allele and 34 on the other (no premutation). Whole exome sequencing was performed as previously described [14] and revealed novel heterozygous variant in a DNA-binding domain of the MYRF gene (RefSeq: NM_001127392.3) – an insertion-deletion c.1318_1319delinsA, causing premature termination of protein synthesis (p.Ala440ThrfsTer2) (Fig. 1C). Heterozygous loss-of-function MYRF variants were described in patients with cardiac-urogenital syndrome (OMIM: 618280) and mild encephalopathy with reversible myelin vacuolization (OMIM: 618113). This variant had neither been reported in association with any phenotype nor reported in the gnomAD database [15]. Sanger sequencing of the patient and her asymptomatic parents was performed and confirmed that the variant arose de novo (Fig. 2). According to the American College of Medical Genetics and Genomics criteria [16], the variant was considered likely pathogenic.
Patient’s data was reconsidered, and her phenotype was in line with MYRF-associated pathology. Therefore, the diagnosis of cardiac-urogenital syndrome was made. The risks for offspring to inherit the pathogenic variant were estimated as 50% independently of sex, according to autosomal dominant inheritance, and preimplantation genetic testing for variant p.Ala440ThrfsTer2 in MYRF gene was recommended to prevent the transmission of the disease.
Discussion
The MYRF gene (MYelin Regulatory Factor) is located on chromosome 11q12. It encodes a transcription factor that plays a significant role in diverse processes. MYRF is involved in central nervous system myelination, eye size regulation and development of heart, lungs, diaphragm, and genitourinary tract. Heterozygous loss-of-function MYRF variants cause multiple congenital anomalies syndrome called cardiac-urogenital syndrome (CUGS). Many patients with CUGS experience developmental delay and intellectual disability [17]. In patients with 46,XY karyotype variants in MYRF cause disorder of sex development, while in some 46,XX patients they lead to genital and gonadal anomalies [18]. Missense variants in MYRF were described in families with mild encephalopathy with reversible myelin vacuolization usually triggered by febrile illness [19].
MYRF is a candidate regulator of early reproductive organs development. It is expressed in a coelomic epithelium during embryonic and fetal stages of ovarian development. MYRF expression is particularly notable in the early developmental stages of the gonadal ridge, which is critical for proper gonadal differentiation. MYRF may regulate other genes involved in gonadal development; for instance, it has been shown to upregulate CITED2, a gene crucial for gonadal differentiation and associated with premature ovarian insufficiency [20]. Three patients with 46,XX karyotype and de novo loss-of-function MYRF variants had ovarian hypoplasia or agenesis and mullerian agenesis, including severely affected patient with congenital diaphragmatic hernia, heart defect and accessory spleen, and one pair of monozygotic twins without extragenital findings [18,21]. In a family with suggested gonadal mosaicism autopsy of a girl with multiple congenital anomalies showed duplication of uterus and vagina, hypoplastic ovaries, splenogonadal fusion, while in sibling autopsy found no urogenital malformations. A splice-site MYRF variant was observed in both siblings, but not in parents [22].
The coincidence of uterus didelphys, ovarian hypoplasia and high hypermetropia and detection of a heterozygous truncating MYRF variant in our patient strongly suggests the diagnosis of a MYRF-related disorder. Vertebral anomalies were not previously described in patients with cardiac-urogenital syndrome, and finding of butterfly vertebrae and kyphoscoliosis in our patient could contribute to the expansion of the phenotypic spectrum of the disease. The patient reported no complaints that could be attributed to mild encephalopathy with reversible myelin vacuolization. The diagnosis of DOR influences reproductive plans of our patient, as it may progress to POI. Additionally, the phenotype of MYRF-associated disorders can be highly variable in males and females even within family members [23]. The possibility of anticipation is illustrated by case of three-generation family, in which the mother of the patient had uterine abnormality, the patient had atrial septal defect, omphalocele and didelphys uterus, and her fetus was diagnosed with isolated hypoplastic left heart syndrome, for which reason the pregnancy was terminated. All three affected subjects carried heterozygous missense variant c.16G>A (p.Glu6Lys) in the MYRF [17]. Therefore, the patient was advised to consider ART with preimplantation genetic testing to avoid the birth of an affected child.
Conclusion
Our case shows pleiotropic action of MYRF transcription factor. Underlying pathogenic variants in the MYRF gene may contribute to abnormal development of both mullerian derivatives and ovaries and diminished ovarian reserve accompanied by other organs and systems dysfunction. It is important to assess ovarian reserve in patients with uterine anomalies to improve reproductive outcomes. Genetic counseling should be provided to patients with uterine anomalies and extragenital pathology or to patients with concomitant uterine anomaly and ovarian hypoplasia, as this combination covers a risk for syndromal genetic pathology.
References
- Chan Y.Y., Jayaprakasan K., Zamora J., Thornton J.G., Raine-Fenning N., Coomarasamy A. The prevalence of congenital uterine anomalies in unselected and high-risk populations: a systematic review. Hum. Reprod. Update. 2011; 17(6): 761-71. https://dx.doi.org/10.1093/humupd/dmr028.
- Cospain A., Dion L., Bidet M., Nyangoh Timoh K., Quelin C., Carton I. et al. Optimizing care for MRKH patients: From malformation screening to uterus transplantation eligibility. Acta Obstet. Gynecol. Scand. 2025; 104(3): 514-21. https://dx.doi.org/10.1111/aogs.14985.
- Chan Y.Y., Jayaprakasan K., Tan A., Thornton J.G., Coomarasamy A., Raine-Fenning N.J. Reproductive outcomes in women with congenital uterine anomalies: a systematic review. Ultrasound Obstet. Gynecol. 2011; 38(4): 371-82. https://dx.doi.org/10.1002/uog.10056.
- Wang Y., He Y.L., Yuan L., Yu J.C., Xue H.D., Jin Z.Y. Typical and atypical pelvic MRI characteristics of Mayer-Rokitansky-Küster-Hauser syndrome: a comprehensive analysis of 201 patients. Eur. Radiol. 2020; 30(7): 4014-22. https://dx.doi.org/10.1007/s00330-020-06681-4.
- Allen J.W., Cardall S., Kittijarukhajorn M., Siegel C.L. Incidence of ovarian maldescent in women with mullerian duct anomalies: evaluation by MRI. AJR Am. J. Roentgenol. 2012; 198(4): W381-5. https://dx.doi.org/10.2214/AJR.11.6595.
- Chen H.A., Grimshaw A.A., Taylor-Giorlando M., Vijayakumar P., Li D, Margetts M., Pelosi E. et al. Ovarian absence: a systematic literature review and case series report. J. Ovarian Res. 2023; 16(1): 13. https://dx.doi.org/10.1186/s13048-022-01090-1.
- Аракелян А.С., Попрядухин А.Ю., Карапетян Э.А. Сочетанные пороки развития в гинекологии. Анализ 1530 клинических наблюдений. (Собственный материал). Российский вестник акушера-гинеколога. 2021; 21(4): 88‑93. [Arakelyan A.S., Popryadukhin A.Yu., Karapetyan E.A. Concomitant malformations in gynecology. Analysis of 1530 clinical observations. (Own material). Russian Bulletin of Obstetrician-Gynecologist. 2021; 21(4): 88-93. (in Russian)]. https://dx.doi.org/10.17116/rosakush20212104188.
- Connell M., Owen C., Segars J. Genetic syndromes and genes involved in the development of the female reproductive tract: a possible role for gene therapy. J. Genet. Syndr. Gene Ther. 2013; 4: 127. https://dx.doi.org/10.4172/2157-7412.1000127.
- Touraine P., Chabbert-Buffet N., Plu-Bureau G., Duranteau L., Sinclair A.H., Tucker E.J. Premature ovarian insufficiency. Nat. Rev. Dis. Primers. 2024; 10(1): 63. https://dx.doi.org/10.1038/s41572-024-00547-5.
- Reyes A.P., León N.Y., Frost E.R., Harley V.R. Genetic control of typical and atypical sex development. Nat. Rev. Urol. 2023; 20(7): 434-51. https://dx.doi.org/10.1038/s41585-023-00754-x.
- Liu S., Gao X., Qin Y., Liu W., Huang T., Ma J. et al. Nonsense mutation of EMX2 is potential causative for uterus didelphysis: first molecular explanation for isolated incomplete müllerian fusion. Fertil. Steril. 2015; 103(3): 769-74.e2. https://dx.doi.org/10.1016/j.fertnstert.2014.11.030.
- Wang X., Zhang X., Liu S., Li G., Cui L., Qin Y. et al. Novel mutations in the TP63 gene are potentially associated with Müllerian duct anomalies. Hum. Reprod. 2016; 31(12): 2865-71. https://dx.doi.org/10.1093/humrep/dew259.
- Li R., Wu S.P., Zhou L., Nicol B., Lydon J.P., Yao H.H. et al. Increased FOXL2 expression alters uterine structures and functions. Biol. Reprod. 2020; 103(5): 951-65. https://dx.doi.org/10.1093/biolre/ioaa143.
- Shubina J., Tolmacheva E., Maslennikov D., Kochetkova T., Mukosey I., Sadelov I. et al. WES-based screening of 7,000 newborns: A pilot study in Russia. HGG Adv. 2024; 5(4): 100334. https://dx.doi.org/10.1016/j.xhgg.2024.100334.
- Chen S., Francioli L.C., Goodrich J.K., Collins R.L., Kanai M., Wang Q. et al.; Genome Aggregation Database Consortium. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. 2024; 625(7993): 92-100. https://dx.doi.org/10.1038/s41586-023-06045-0.
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J. et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015; 17(5): 405-24. https://dx.doi.org/10.1038/gim.2015.30.
- Favier M., Brischoux-Boucher E., Pyle L.C., Mottet N., Auber-Lenoir M., Cattin J. et al. Fetal presentation of MYRF-related cardiac urogenital syndrome: an emerging and challenging prenatal diagnosis. Prenat. Diagn. 2024; 44(13):1647-58. https://dx.doi.org/10.1002/pd.6700.
- Hamanaka K., Takata A., Uchiyama Y., Miyatake S., Miyake N., Mitsuhashi S. et al. MYRF haploinsufficiency causes 46,XY and 46,XX disorders of sex development: bioinformatics consideration. Hum. Mol. Genet. 2019; 28(14): 2319-29. https://dx.doi.org/10.1093/hmg/ddz066.
- Kurahashi H., Azuma Y., Masuda A., Okuno T., Nakahara E., Imamura T. et al. MYRF is associated with encephalopathy with reversible myelin vacuolization. Ann. Neurol. 2018; 83(1): 98-106. https://dx.doi.org/10.1002/ana.25125.
- Calonga-Solís V., Fabbri-Scallet H., Ott F., Al-Sharkawi M., Künstner A., Wünsch L. et al. MYRF: A new regulator of cardiac and early gonadal development-insights from single cell RNA sequencing analysis. J. Clin. Med. 2022; 11(16): 4858. https://dx.doi.org/10.3390/jcm11164858.
- Qi H., Yu L., Zhou X., Wynn J., Zhao H., Guo Y. et al. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet. 2018; 14(12): e1007822. https://dx.doi.org/10.1371/journal.pgen.1007822.
- Slaba K., Jezova M., Pokorna P., Palova H., Tuckova J., Papez J. et al. Two sisters with cardiac-urogenital syndrome secondary to pathogenic splicing variant in the MYRF gene with unaffected parents: A case of gonadal mosaicism? Mol. Genet. Genomic Med. 2023; 11(5): e2139. https://dx.doi.org/10.1002/mgg3.2139.
- Gupta N., Endrakanti M., Gupta N., Dadhwal V., Naini K., Manchanda S. et al. Diverse clinical manifestations and intrafamilial variability due to an inherited recurrent MYRF variant. Am. J. Med. Genet. A. 2022; 188(7): 2187-91. https://dx.doi.org/10.1002/ajmg.a.62744.
Received 16.12.2024
Accepted 03.04.2025
About the Authors
Polina N. Tsabai, geneticist, M.D., Department of Clinical Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, polinatsabai@gmail.com, https://orcid.org/0000-0001-5110-0827Zalina K. Batyrova, gynecologist, M.D., PhD., Department of Pediatric and Adolescent Gynecology, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, linadoctor@mail.ru, https://orcid.org/0000-0003-4997-6090
Zaira Kh. Kumykova, gynecologist, M.D., PhD., Department of Pediatric and Adolescent Gynecology, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, zai-kumykova@yandex.ru,
https://orcid.org/0000-0001-7511-1432
Valeria A. Kovalskaia, geneticist, M.D., Department of Clinical Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, mikhailova.v.a@mail.ru, https://orcid.org/0000-0002-8728-8574
Mariia A. Sidorchuk, geneticist, M.D., Department of Clinical Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, sidorchuk.mariia.98@gmail.com, https://orcid.org/0009-0008-1373-7905
Andrey Yu. Goltsov, researcher, Laboratory of Molecular Genetic Methods, Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, andrey.goltsov@gmail.com,
https://orcid.org/0000-0002-4004-4214
Igor O. Sadelov, geneticist at the Laboratory for the Genomic Data Analysis, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, sadelovigor@gmail.com, https://orcid.org/0000-0001-5916-0672
Ekaterina R. Tolmacheva, researcher at the Laboratory for the Genomic Data Analysis, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, +7(903)135-95-48, tetisae@gmail.com,
https://orcid.org/0000-0003-2901-0539
Elena V. Uvarova, Dr. Med. Sci., Professor, Corresponding Member of RAS, Head of Department of Pediatric and Adolescent Gynecology, Academician V.I. Kulakov
National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997,
elena-uvarova@yandex.ru, https://orcid.org/0000-0002-3105-5640
Gennady T. Sukhikh, Dr. Med. Sci., Professor, Academician of RAS, Director, Academician V.I. Kulakov National Medical Research Center for Obstetrics,
Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, https://orcid.org/0000-0002-7712-1260
Dmitry Yu. Trofimov, Dr. Med. Sci., Professor of the RAS, Corresponding Member of RAS, Director of the Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, molgen@bk.ru, https://orcid.org/0000-0002-1569-8486
Corresponding author: Polina N. Tsabai, polinatsabai@gmail.com
Similar Articles

