
Синдром делеции хромосомы 22 (22q11.2 deletion syndrome, 22q11DS) представляет совокупность морфологических, иммунологических и неврологических изменений вследствие потери длинного плеча 1 копии 22-й хромосомы и является наиболее часто встречаемой из числа всех аутосомных микроделеционных синдромов и второй, после трисомии 21, хромосомной причиной формирования врожденного порока сердца (ВПС) [1–3].
Значительная часть (80–90%) пациентов с синдромом 22q11DS имеют типичную гемизиготную делецию размером около 3 млн пар нуклеотидов; у 7% пациентов выявляют делецию меньших размеров (около 1,5 млн пар нуклеотидов; в незначительном проценте случаев 22q11DS обнаруживают атипичную делецию) [4–6].
Предполагаемая распространенность 22q11DS соответствует 1 на 3000–6000 живорождений и около 1 на 1000 плодов [1, 3]. Указанная структурная патология хромосомы 22 с одинаковой частотой встречается у обоих полов [7]. При этом необходимо отметить, что, по данным значительного числа публикаций, в 90% случаев и более делеция происходит de novo [1–3]. Однако в одном из крупнейших пренатальных исследований Besseau-Ayasse J. et al. (2014), изучавшем тип наследования данной делеции у 189 плодов с 22q11DS, была установлена частота семейных форм передачи, равная 27%, при этом в 76,1% наблюдений наследование происходило по материнской линии [8]. При семейных формах рассматриваемая делеция наследуется по аутосомно-доминантному типу [9, 10] с риском повторения 50%, в то время как при отсутствии генетической патологии у обоих родителей риск повторения 22q11.2DS у следующего ребенка не превышает 1% [11].
Клинические проявления делеции хромосомы 22q11.2DS обусловлены дефектом развития 3-й и 4-й фарингеальных дуг и 4-й дуги аорты в периоде эмбриогенеза, приводящим к широкому спектру мультисистемных поражений [12]. Синдром del22q11, ранее также называемый САТСН 22 (ВПС (Cardiac defects), аномальное лицо (Abnormal facies), гипоплазия тимуса (Thymic hypoplasia), расщелина неба (Cleft palate), гипокальциемия (Hypocalcemia), del 22), связан с очень вариабельным фенотипом, включающим более 100 различных клинических проявлений. При этом необходимо отметить, что в ряде отечественных и зарубежных публикаций в структуре 22q11DS исследователи выделили отдельные клинические синдромы: синдром Ди Джорджи (МКБ-10 D82.1 Синдром Ди Георга, ОМIM #188400 DiGeorge syndrome, DGS), велокардиофациальный синдром (ОМIM #192430, Velocardiofacial syndrome, VCFS), синдром конотрункальных и лицевых аномалий (Conotruncal anomalies face syndrome, CTAF), синдром Кайлера (Cayler cardiofacial syndrome) [1, 12]. Известна клиническая вариабельность проявлений 22q11.2DS даже у пациентов с аналогичными пограничными точками и размером делеции, в том числе в пределах одной семьи [13, 14].
Наиболее распространенные признаки 22q11DS включают лицевой дисморфизм: монголоидный разрез глаз, гипертелоризм, узкую ротовую щель, низко посаженные уши, ретрогнатию и патологию неба, приводящие к нарушению речи и расстройствам питания, иммунодефицит и рецидивирующие инфекции, гипокальциемию и судороги, нарушения развития нервной системы (задержку развития и психические нарушения, включая шизофрению), а также ВПС [1, 15].
ВПС являются не только одними из наиболее частых, но и самыми жизнеугрожающими из проявлений синдрома 22q11DS: структурные аномалии сердца и сосудов встречаются у 60–80% пациентов [1, 8] и обуславливают 87% летальных исходов у детей [12]. Данные Goldmuuntz E. [1] демонстрируют, что, несмотря на то, что у пациентов с 22q11DS не отмечается достоверно более высокой смертности при коррекции ВПС, они сталкиваются со значимо большим количеством периоперационных осложнений по сравнению с больными без генетических нарушений, перенесшими аналогичное по объему хирургическое вмешательство [1]. Так, Alsoufi B. et al. (2017) [16] при анализе исходов оперативного лечения новорожденных с ВПС установили, что по сравнению с детьми без генетических нарушений в группе пациентов с делецией 22q11.2DS значимо больше была продолжительность искусственной вентиляции легких, длительность пребывания в отделении интенсивной терапии и госпитализации в целом. В проспективном датском национальном наблюдении за когортой взрослых, оперированных ранее по поводу тетрады Фалло или атрезии легочной артерии, Kauw D. et al. (2020) [17] было подтверждено, что группа пациентов с наличием 22q11.2DS имела значительно худшую выживаемость в сравнении с больными без указанной делеции. Авторы указывают на то, что даже у взрослых пациентов с данными типами ВПС необходимо генетическое тестирование на наличие 22q11.2DS для стратификации риска развития послеоперационных осложнений [17].
В большинстве наблюдений именно клиническая манифестация ВПС на пре- или постнатальном этапе является отправной точкой в диагностике генетической аномалии. Campbell I.M. et al. (2018) [7] в одном из наиболее крупных лонгитудинальных исследований, посвященных наблюдению за когортой из 1421 пациента с подтвержденной делецией хромосомы 22q11DS, отметили, что при наличии ВПС диагноз синдромальной патологии у ребенка был установлен значительно раньше – медианный возраст 2,6 месяца в сравнении со случаями без ВПС – медианный возраст 3,1 года [7].
К ВПС, наиболее часто встречающимся при 22q11.2DS, относят тетраду Фалло (20–45% пациентов), атрезию легочной артерии (10–20% пациентов), перерыв дуги аорты, преимущественно тип В, (5–20% пациентов), общий артериальный ствол (ОАС) (5–10% пациентов), дефекты конусной перегородки (10–50% пациентов) [3, 12, 18]. Также могут быть обнаружены двойной выход магистральных сосудов из правого желудочка, транспозиция магистральных артерий, прочие аномалии дуги аорты [3, 12, 18].
На современном этапе развития ультразвуковой диагностики все вышеперечисленные аномалии развития непременно должны быть выявлены при пренатальном обследовании. Основным эхографическим маркером высокого риска наличия 22q11DS у плода с ВПС является уменьшение тимо-торакального отношения (ТТО) как проявление аплазии либо гипоплазии тимуса [19–21]. Несмотря на то что существует общепринятый способ расчета ТТО как отношения переднезаднего размера тимуса к интраторакальному размеру в стандартном срезе через 3 сосуда и трахею [19], до настоящего времени однозначного консенсуса о референсных интервалах данного показателя не достигнуто. В большинстве проведенных исследований было показано, что средние значения ТТО во второй половине беременности у здоровых плодов были равны 0,41–0,44 и значительно превышали таковые у плодов с 22q11DS [19–22]. Наряду с этим рядом исследований показано, что снижение ТТО нередко встречается у плодов с конотрункальными ВПС, не имеющими структурных изменений 22q11DS [21, 22]. При этом снижение ТТО у этой группы пациентов четко ассоциировалось с худшим прогнозом хирургического лечения новорожденных [21, 22]. Также было показано, что фактором, существенно увеличивающим риск наличия 22q11DS у плода, является сочетание уменьшения значений ТТО с отклонением оси сердца влево [19, 23].
Возможность экстраполирования разработанных на когортах плодов при одноплодной беременности эхографических критериев на случаи многоплодия подтверждена результатами исследования Gamez F. et al. (2010) [24], в котором не было выявлено статистически значимой разницы размеров тимуса здоровых плодов как при одноплодной, так и при двуплодной беременности.
В общей популяции беременных особую настороженность в отношении аномалий развития сердечно-сосудистой системы у плода должны вызывать случаи монохориального многоплодия, поскольку накопленный опыт наблюдений демонстрирует шестикратное повышение частоты ВПС у данной группы по сравнению с одноплодной беременностью. Кроме этого, необходимо отметить, что особую настороженность требуют плоды при развитии фето-фетального трансфузионного синдрома, при котором у выживших плодов частота патологии сердца дополнительно двукратно увеличивается [25, 26].
Несомненный клинический и научный интерес представляют те факты, что именно ВПС являются самыми частыми из всех мальформаций у плодов из монохориальных двоен и что в более чем 95% случаев указанные пороки развития встречаются только у одного из плодов [25, 27, 28]. При этом показано, что частота генетической патологии при ВПС существенно ниже при монохориальном, чем при дихориальном типе многоплодия. Так, в исследовании Zhang Y. at al. (2018) [28], включавшем 149 двоен с ВПС у одного либо обоих плодов, аномальные результаты кариотипирования и хромосомного микроматричного анализа были получены в 7,7 и 21,3% случаев при монохориальных двойнях, а также в 18,3 и 32,1% – при дихориальных двойнях соответственно. Аналогичные выводы были сделаны и по результатам работы Li L. et al. (2020) [27], которые на когорте из 152 плодов с ВПС установили меньшую частоту аномалий кариотипа при монохориальных, чем при дихориальных двойнях: 16,0 и 39,2% соответственно.
Несмотря на предполагаемую значительную частоту 22q11.2DS среди плодов с ВПС [1, 16], данные о пренатальном выявлении указанной патологии у плодов при многоплодной беременности представлены только в результатах масштабного исследования обсуждаемого синдрома, проведенного Sivrikoz T.S. et al. (2022) [14]. В когорте данного исследования, включающей 48 плодов с подтвержденным диагнозом 22q11.2DS, была только одна дихориальная двойня, дискордантная по наличию ВПС: коарктация аорты была диагностирована у одного плода из двойни; структурной патологии у второго плода пренатально выявлено не было. Беременность была прервана по желанию родителей. Постмортальное исследование выявило фенотипические особенности строения лица и постаксиальную полидактилию одной из кистей у обоих плодов. Проведенный генетический анализ подтвердил монозиготность двойни и наличие у обоих плодов 22q11.2DS с делецией, возникшей de novo [14].
Осуществленный скрупулезный анализ литературных источников позволил обнаружить 9 опубликованных случаев постнатальной диагностики 22q11DS у пар монозиготных близнецов [10, 29–35]. Особенности кардиальной и сопутствующей патологии, а также отдаленные исходы каждой пары представлены в таблице. При этом необходимо отметить, что семейная форма наследования была определена лишь в одном случае [10].
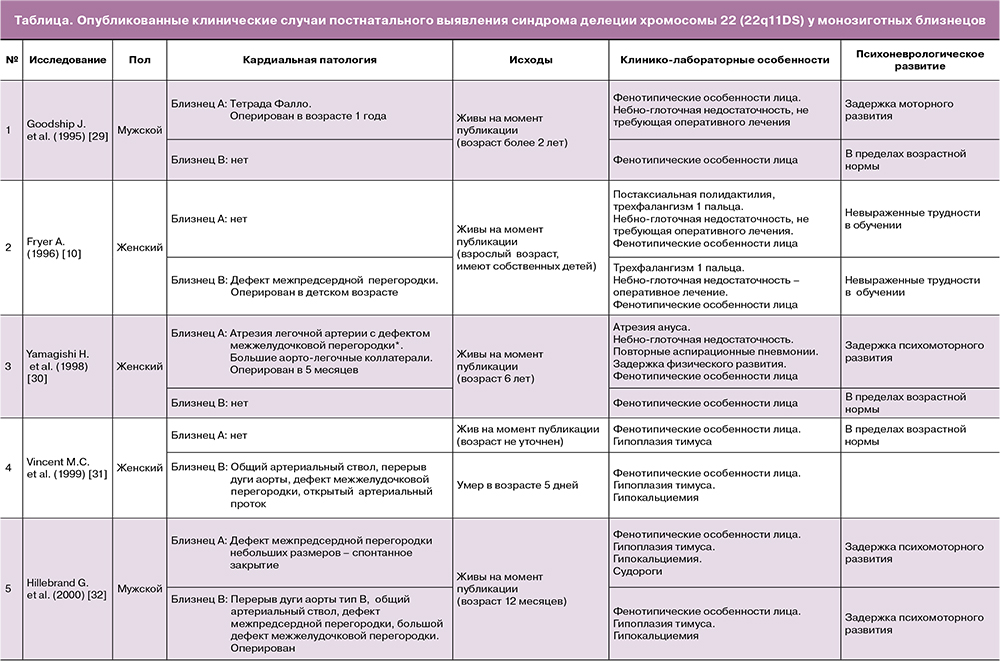
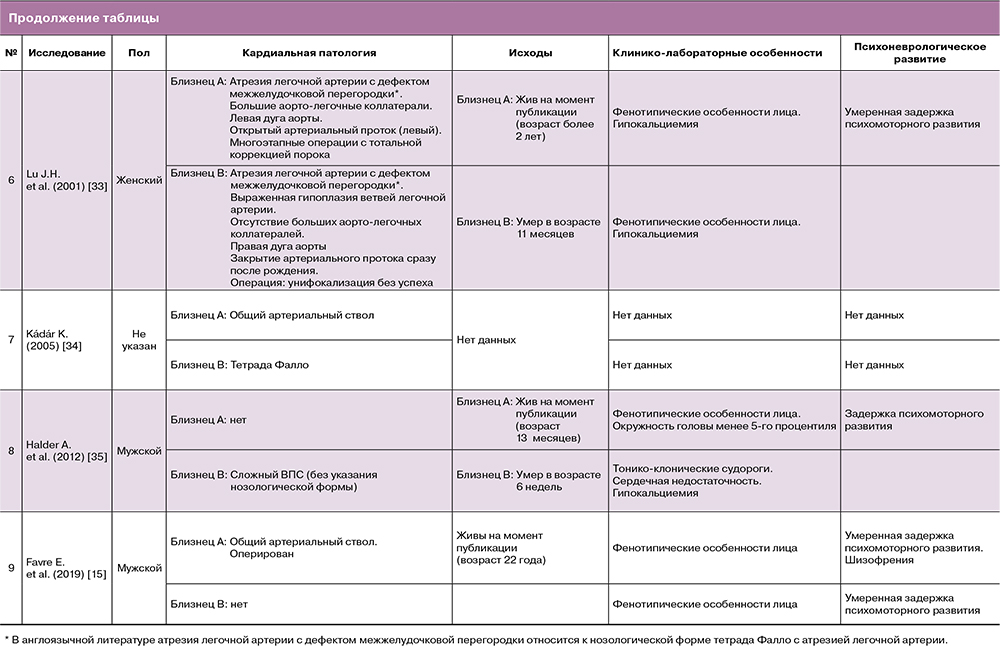
Из общей когорты 18 близнецов неизмененная кардиальная анатомия была констатирована у 6 детей, у 2 выявлены дефекты межпредсердной перегородки, не требующие оперативного лечения, и у 10 новорожденных – более сложные ВПС: у 4 – ОАС, у 3 – атрезия легочной артерии с дефектом межжелудочковой перегородки, у 2 – тетрада Фалло и у 1 ребенка – сложный ВПС, нозологическая форма которого точно не установлена.
Важно отметить, что во всех представленных наблюдениях не что иное, как манифестация кардиальной патологии у одного плода из двойни, послужила основанием для проведения диагностического поиска, клинического и лабораторного обследования, позволившего установить наличие генетической патологии. Также во всех случаях отмечалась дискордантность патологических изменений анатомии сердечно-сосудистой системы близнецов: от полного отсутствия структурной патологии до сложных ВПС, которые привели к летальному исходу ребенка до или после операции [31, 33, 35]. Данный факт подтверждается исследованием Karbarz M. (2020) [13], в котором отмечено, что делеция хромосомы 22q11.2DS – показательный пример непредсказуемого фенотипа, так как, несмотря на обширные генетические познания и высокоточные генетические тесты, до настоящего времени не существует однозначного объяснения, почему монозиготные близнецы с точно такой же делецией имеют разные фенотипы.
Именно дискордантностью выраженности гипоплазии системы легочных артерий в случае, описанном Lu J.H. et al. (2001) [33], была обусловлена неудачная попытка хирургической коррекции порока у одного из пары близнецов с, казалось бы, конкордантной патологией: атрезией легочной артерии.
В общей сложности из 8 пар близнецов, для которых описаны отдаленные результаты, на первом году жизни погибли 3 ребенка со сложными пороками сердца [31, 33, 35].
Особого внимания в контексте различных нозологических форм ВПС при 22q11.2DS также заслуживают данные, представленные Fryer A. (1996) [10] с семейным случаем близнецов-женщин, не имевших сложной кардиальной патологии: только одной из них потребовалось кардиохирургическое лечение по закрытию дефекта межпредсердной перегородки. Однако впоследствии в данной семье произошла передача патологической делеции 22-й хромосомы следующему поколению, в котором один из трех детей – носителей 22q11.2DS – погиб после проведения хирургической коррекции сложного ВПС: гемитрункус (отхождение левой легочной артерии от ветвей аорты) и отсутствие клапана легочного ствола с тяжелой легочной регургитацией [10].
Из 13 детей с 22q11.2DS, у которых представлены сведения их последующего развития, только у 3, не имеющих кардиальной патологии, не было отмечено патологических психоневрологических либо моторных нарушений [29–31].
На современном этапе развития дородовой ультразвуковой диагностики, включающей пренатальную эхокардиографию, очевидна возможность антенатальной диагностики ВПС, ассоциированных с делецией хромосомы 22q11.2DS, в том числе и у монохориальных двоен – популяции плодов, которым показано проведение расширенного и прицельного эхокардиографического исследования [36].
Однако на момент написания статьи нашему авторскому коллективу не удалось обнаружить ни в отечественных, ни в зарубежных журналах опубликованные данные о пренатальном выявлении ВПС у плодов со структурными изменениями хромосомы 22q11DS при монохориальной беременности, что делает чрезвычайно интересным представление следующего клинического наблюдения.
Клиническое наблюдение
Нами представляется клиническое наблюдение пациентки К. 30 лет, впервые обратившейся в Перинатальный кардиологический центр ФГБУ «НИМЦ ССХ им. А.Н. Бакулева» Минздрава России в сроке беременности 20 недель 5 дней. В анамнезе у пациентки отмечены перенесенные детские инфекции, ОРВИ, удаление фиброаденомы правой молочной железы в возрасте 28 лет. Гинекологический анамнез не отягощен. Пациентка состоит в первом браке. Возраст супруга – 35 лет, соматически здоров. Вредные привычки оба родителя отрицают. Семейный анамнез не отягощен с обеих сторон. Данная беременность первая, наступила естественным путем. При ультразвуковом исследовании в сроке 8 недель была диагностирована многоплодная беременность – монохориальная диамниотическая двойня.
Скрининговые исследования I и II триместра были проведены по месту амбулаторного наблюдения пациентки. Комбинированный скрининг I триместра не выявил эхографических признаков врожденных пороков развития и хромосомных аномалий у плодов. Риск трисомии 21, 18 и 13 у обоих плодов определен, как низкий. При проведении ультразвукового исследования в сроке 19 недель беременности была выявлена аномальная эхографическая картина при сканировании через четырехкамерный срез сердца и срез через магистральные сосуды у обоих плодов, предварительно сформулированы диагнозы ВПС: тетрада Фалло у первого плода, ОАС – у второго плода.
По причине желания семейной пары пролонгировать беременность в связи с наличием возможности постнатальной хирургической коррекции данного типа пороков пациентка была направлена в Перинатальный кардиологический центр для проведения экспертной пренатальной эхокардиографии и консультации кардиохирурга с целью уточнения нозологической формы ВПС и прогноза для жизни и здоровья детей. Ультразвуковое исследование было проведено на системе экспертного класса Voluson E8 Expert (GE Healthcare, Австрия) с использованием 3D/4D трансабдоминального конвексного мультичастотного (2–8 MHz) датчика. При проведении исследования констатировано недиссоциированное развитие плодов, соответствие размеров обоих плодов сроку беременности, неизмененное количество околоплодных вод у обоих плодов, а также не было выявлено признаков фето-фетального трансфузионного синдрома. Допплерографические показатели кровотока в маточных артериях, артериях пуповин и средних мозговых артериях плодов находились в пределах нормативных гестационных значений.
При проведении эхокардиографии у обоих плодов определялась аналогичная эхографическая картина: неизмененное положение органов брюшной полости и грудной клетки (situs solitus), нормальные размеры сердца, отклонение оси сердца влево более чем на 75 градусов. Четырехкамерная проекция была не изменена, определялись нормальное венозно-атриальное и конкордантное предсердно-желудочковое соединение, размеры и положение предсердий и желудочков соответствовали гестационной норме. Проекция выходных трактов левого/правого желудочков была аномальной. Определялся единственный артериальный сосуд (трункус – ОАС) с бивентрикулярным отхождением за счет наличия подтрункального дефекта межжелудочковой перегородки, отхождение ствола легочной артерии от боковой стенки трункуса (рис. 1) с последующим делением его на правую и левую легочные артерии. При цветовой допплерографии определялась выраженная недостаточность трункального клапана, антеградный кровоток в легочной артерии (рис. 1). Проекция трех сосудов и трахеи также была аномальной – вместо трех сосудов визуализировались только два: нормального диаметра верхняя полая вена и сосуд большего диаметра – трункус. Типичного V-соединения сосудов получить не удалось. Артериальный проток не визуализировался в типичном месте. Сагиттальный срез дуги аорты был не изменен – нормального диаметра брахиоцефальные сосуды отходили от дуги аорты с нормальным типом ветвления. ТТО было уменьшено до 0,18 у первого и до 0,21 – у второго плода (рис. 1). Признаков патологии со стороны других органов и структур обоих плодов не было выявлено.

Окончательный пренатальный диагноз был сформулирован следующим образом: Многоплодная беременность сроком 20 недель 5 дней с учетом даты первого дня последнего менструального цикла: монохориальная диамниотическая двойня. ВПС у обоих плодов: ОАС I типа по классификации Van Praagh. Уменьшение ТТО у обоих плодов. Высокий риск синдромальной патологии плодов (включая синдром делеции хромосомы 22).
Проведены консультация кардиохирурга и перинатальный консилиум, определившие неблагоприятный прогноз для жизни и развития обоих плодов. Пациентка информирована о целесообразности проведения генетического исследования для определения причины развития ВПС у плодов и оценки прогноза для будущего потомства. По решению семьи беременность была прервана в стационаре по месту проживания пациентки, подтвержден монохориальный тип многоплодия и ВПС у обоих плодов.
Фрагменты плацентарной ткани доставлены в Институт репродуктивной генетики ФГБУ «Национальный медицинский исследовательский центр акушерства, гинекологии и перинатологии им. академика В.И. Кулакова» Минздрава России, где проведен хромосомный микроматричный анализ ДНК плацентарной ткани (с использованием микроматриц Cytoscan Optima Array), выявивший генетическую патологию. Результаты Cytoscan arr[hg19] 22q11.21(18,917,030-21,465,662) x1: в анализируемом материале с мужским генотипом выявлена патогенная микроделеция на длинном плече хромосомы 22 размером более 2,5 млн пар нуклеотидов с вовлечением 41 гена, описанного в базе данных OMIM (в том числе TBX1). Рекомендованы очная консультация врача-генетика, а также генетическое обследование семейной пары.
ОАС (OMIM # 217095) является редким ВПС с общей частотой 0,03–0,056 на 1000 живорожденных, что составляет 0,21–0,34% всех случаев врожденных пороков сердца. В Европейских странах средняя зарегистрированная распространенность составляет 1 случай на 10 000 беременностей (включая живорождения, мертворождения и прерывания беременности) [37]. До настоящего времени только в зарубежной литературе опубликованы несколько случаев постнатального выявления ОАС у обоих близнецов, все они не были ассоциированы с делецией хромосомы 22q11.2DS [38–40]. В российских публикациях подобных данных не представлено, что подтверждает особую актуальность представленного нами клинического наблюдения.
Морфогенетически основные черты ОАС можно отнести к аномалиям развития аортопульмонального септального комплекса, приводящего к отсутствию аортопульмональной перегородки на 3 уровнях: уровне магистральных артерий, клапанном уровне и уровне выходных трактов [41]. Без оперативного лечения данный порок относится к фатальным. Вместе с тем, благодаря современным методам диагностики и хирургической техники, периоперационная летальность среди детей может быть снижена до 5–10%, а выживаемость более 30 лет может достигать 73,6%, при высокой частоте повторных вмешательств, наиболее часто на выходном тракте правого желудочка [41].
Однако статистические данные и приведенные клинические случаи свидетельствуют о том, что, несмотря на успехи кардиохирургии, при пренатальном консультировании семейной пары необходимо помнить о значительной частоте ассоциации ОАС с 22q11.2DS [18]. Согласно данным зарубежных публикаций, 22q11.2DS выявляется в 35% случаев наличия ОАС у плода [41], что также подтверждено результатами Новиковой И.В. и соавт. (2020) [42], проводивших патоморфологическое исследование плодов с 22q11.2DS и установивших, что ОАС в сочетании с гипоплазией тимуса относятся к одной из наиболее частых патологических находок. В литературных источниках имеются указания на то, что гипоплазия тимуса может быть ассоциирована с высоким риском неонатального сепсиса и неблагоприятным прогнозом при хирургической коррекции порока сердца [1, 19]. Так, по данным Дерябиной С.С. и соавт. (2020) [43], диагноз генетически обусловленного первичного иммунодефицита при ВПС у ребенка чаще всего выставляется постмортально. Авторами постулировано, что именно увеличение осведомленности клинических специалистов о возможностях генетической диагностики у плодов с ВПС может приводить к улучшению качества помощи детям с первичной иммунной недостаточностью, отягощенной сердечно-сосудистой патологией [43].
При любом типе ВПС крайне важно на пренатальном этапе выявить сопутствующую экстракардиальную патологию [44, 45], наличие которой негативно влияет не только на продолжительность, но и на качество жизни как самого пациента, так и его семьи. При этом на пренатальном консультировании необходимо учитывать также существующие ограничения возможностей ультразвукового исследования в выявлении всего спектра имеющейся врожденной патологии органов и систем плода. Так, Besseau-Ayasse J. et al. (2014) [8] сравнили результаты патологоанатомического и ультразвукового исследований 83 плодов с 22q11.2DS и установили, что пренатально аномалии тимуса были выявлены только у 4 из 44 плодов, патология почек – у 11 из 22, расщелины неба – у 3 из 4 плодов.
В отношении 22q11.2DS немаловажно отметить, что, помимо мультисистемных соматических поражений, данный синдром относится к одному из крупнейших известных генетических факторов риска развития шизофрении [46, 47]. Распространенность последней в течение жизни составляет примерно 25% при делеции хромосомы 22q11.2DSпо сравнению, с примерно 1% в общей популяции [48]. Структурные изменения 22q11.2DS также проявляются в виде различных психоневрологических симптомов, таких как когнитивная дисфункция, расстройство аутистического спектра или синдром дефицита внимания с гиперактивностью [46, 48]. Широкий спектр экстракардиальной соматической и психоневрологической патологии отмечен в значительном проценте приведенных в данном обзоре клинических наблюдений монозиготных двоен с делецией хромосомы 22q11.2DS [10, 29–35].
Представленные выше данные свидетельствуют о том, что при установлении монохориального многоплодия в обязательном порядке необходимо направлять пациентку для проведения в 20 недель беременности экспертной эхокардиографии плодов [36]. Все клинические специалисты, оказывающие медицинскую помощь беременным женщинам, должны быть знакомы с проявлениями синдрома делеции хромосомы 22 у плода, и при возникновении подозрений безотлагательно направлять пациентку на молекулярно-генетическое исследование.
Диагностика 22q11DS в настоящее время может производиться с помощью различных лабораторных методов: флуоресцентной гибридизации in situ (FISH), мультиплексной амплификации лигированных олигонуклеотидов (MLPA), хромосомного микроматричного анализа (ХМА), высокопроизводительного секвенирования (NGS) [4–6, 49–59]. MLPA и FISH предназначены для таргетной диагностики частых генетических синдромов, что ограничивает их применение в случае выявления кардиальной патологии плода [49–51]. ХМА позволяет выявлять не только анеуплоидии, крупные делеции и дупликации, но также и субмикроскопические перестройки хромосом [51]. Благодаря этому в пренатальной диагностике у плодов с аномалиями развития ХМА показал дополнительную диагностическую ценность порядка 6% по сравнению со стандартным цитогенетическим исследованием [52–54]. В 2013 г. Американским обществом акушеров- гинекологов и Обществом медицины матери и плода были выпущены рекомендации, согласно которым у плодов с аномалиями развития ХМА может заменить или дополнить анализ кариотипа [55–57]. Применение при проведении ХМА SNP-олигонуклеотидных микроматриц позволяет обнаружить не только хромосомные микроделеции/микродупликации, но и однородительские изодисомии, а также большинство полиплоидий [57, 58]. Преимуществом метода является возможность работы с некультивированными клетками околоплодных вод, плаценты, биоптатов хориона и тканей плода [53], что позволяет получать результаты в короткие сроки.
Максимально раннее – на дородовом этапе – выявление синдрома делеции хромосомы 22 у плода позволяет не только установить причину формирования ВПС, но и прогнозировать развитие экстракардиальных соматических, иммунологических и нервно-психических нарушений. Указанные обстоятельства позволяют провести пренатальное консультирование семейной пары, предоставив родителям информированный выбор в отношении пролонгирования беременности, а также скорректировать лечение ребенка сразу после рождения.
Выявление 22q11.2DS у плодов или детей является показанием для генетического обследования родителей для уточнения характера наследования заболевания, поскольку семейные формы заболевания могут не иметь фенотипических проявлений в каждом из поколений [9].
На основании изученных литературных данных нами разработан алгоритм обследования и консультирования, направленный на выявление синдрома делеции 22q11.2 у плода и родителей при одноплодной беременности и различных типах многоплодия (рис. 2).
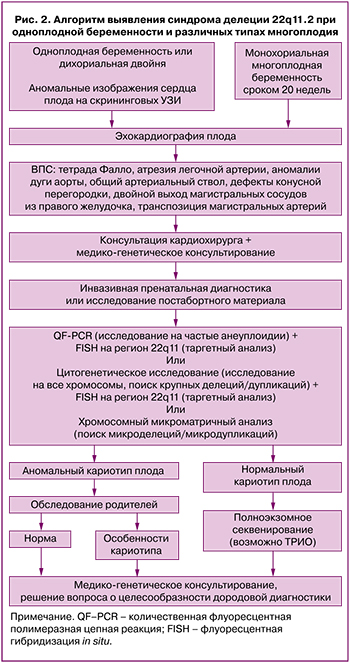
В случаях, когда родитель является носителем хромосомной перестройки, для снижения вероятности рождения больного ребенка может быть рекомендовано проведение экстракорпорального оплодотворения с преимплантационной генетической диагностикой [59]. При спонтанно наступившей беременности рекомендована расширенная пренатальная диагностика, включающая экспертное ультразвуковое исследование плода, эхокардиографию (позволяющие обнаружить уже с 11–13 недель беременности врожденные пороки развития различных органов и систем плода, включая такие сложные ВПС, как тетрада Фалло, ОАС, атрезия легочной артерии) и инвазивную генетическую диагностику. Выявление генетической патологии и ВПС у плода в I триместре предоставляет матери больше возможностей для принятия решения относительно пролонгирования беременности и сохранения своего физического и психологического здоровья.
Значимость всего вышеперечисленного в буквальном смысле двукратно возрастает в случаях монохориальных двоен, когда на чаши весов положены две жизни и благополучие семьи, заботящейся о двух неизлечимо больных детях.
Заключение
Таким образом, в данной публикации представлено собственное, первое в Российской Федерации, клиническое наблюдение пренатального выявления конкордатного ВПС – ОАС у плодов в монохориальной двойне. Благодаря обстоятельной оценке дополнительных ультразвуковых критериев, таких как ТТО и угол сердечной оси, в сроке 20–21 неделя беременности был определен высокий риск наличия делеции хромосомы 22q11.2DS, подтвержденой впоследствии при исследовании плацентарной ткани. Приведен обзор литературы, посвященной синдрому делеции 22-й хромосомы при монозиготных двойнях. Детально проанализирован весь спектр кардиальной и сопутствующей патологии, клиническое течение и отдаленные исходы в литературных источниках. Предложен диагностический алгоритм, клиническое применение которого должно обеспечить максимально раннее выявление 22q11.2DS плодов с ВПС и всестороннее консультирование с формированием репродуктивного прогноза для семейной пары.


