
Врожденные диафрагмальные грыжи (ВДГ) являются одной из наиболее часто встречающихся аномалий развития у новорожденных – в среднем 1/2000–3000 родов. Без экстренной хирургической помощи большая часть детей с ВДГ погибает в первые часы жизни. Согласно данным Росстата, «Врожденные аномалии (пороки развития), деформации и хромосомные нарушения» (Q00-Q99 МКБ-10) занимают второе место среди причин перинатальной смертности в Российской Федерации, составляя в 2010 году 4,7% всех случаев мертворождения [1].
Пренатальная диагностика пороков развития вообще и диафрагмальной грыжи в частности не всегда бывает успешной на ранних сроках беременности (до достижения плодом возраста жизнеспособности). Большинство случаев ВДГ (67%) удается диагностировать только в конце II–III триместрах беременности [2–5].
ВДГ является весьма гетерогенной группой аномалий развития по своим анатомическим, морфологическим и генетическим характеристикам. При сочетании ДГ с другими аномалиями развития патология чаще имеет установленную генетическую этиологию – хромосомную и моногенную. Среди хромосомных синдромов ДГ встречает при трисомии 13 (синдром Патау), трисомии 18 (синдром Эдвардса), делеции короткого плеча хромосомы 4 (синдром Вольфа–Хиршхорна), тетрасомии по короткому плечу хромосомы 12 (синдром Паллистера–Киллиана), а также при более редких вариантах хромосомных аберраций – del 1q, 3q, 8p, 8q, 15q, dup 1q,2p,4q, 22q [6–8].
Среди моногенных форм множественных аномалий развития, включающих ДГ, наиболее известны аутосомно-доминантные синдромы Cornelii de Lange, Kabuki [9], аутосомно-рецессивные синдромы – Fryns, летальный синдром множественных птеригиумов [10], синдром Donnai–Barrow [11, 12], а также X-сцепленные рецессивные синдромы (пентада Кантрелла, Симпсона–Голаби и ряд других).
Для многих генных и хромосомных синдромов, включающих в себя ДГ, современные методы молекулярного анализа позволили установить первичный генный дефект [12].
Изучается роль врожденных моногенных аутосомно-доминанатных дефектов соединительной ткани, которые могут приводить к развитию ДГ в частности, синдрома Марфана, Элерса–Данлоса [13, 14]. Более того, установлено наличие локусов- кандидатов, ответственных за формирование дефектов соединительной ткани, в том числе и ДГ. При синдроме Марфана обсуждается роль мутации FBN1 [14]. Описан синдром микроделеции 15q24, при котором наблюдается системное поражение соединительной ткани с формированием ДГ [15].
Особый интерес представляют собой Х-сцепленные генетические синдромы, включающие с себя ВДГ. Известно преобладание среди больных с данной патологией лиц мужского пола. По данным литературы [16, 17], гендерное соотношение при ДГ составляет 2 мальчика : 1 девочка. Установлено наличие мутаций рядя генов, локализованных на Х хромосоме при пентаде Кантрелла Хq25-26.1, Х-сцепленном синдроме MIDAS (микрофтальмия с линейными дефектами кожи) [18], мутация на коротком плече Х-хромосомы в виде микродупликации Xq12q13.1, затрагивающей ген EFNB1 [19].
Изолированные односторонние ДГ, как правило, имеют мультифакториальную природу. Предполагается влияние средовых тератогенных факторов в период формирования купола диафрагмы на 5–8 неделях эмбрионального развития.
 В экспериментах на животных показано влияние ретинола на процесс возникновения ВДГ и гипоплазии легких за счет супрессии гена Midkine, участвующего в выработке фактора роста легочной ткани (RA-responsive growth factor) [20–22]. Роль уровня ретинола в развитии ВДГ подтверждена при исследовании новорожденных и их матерей [23]. Также обсуждается влияние диабета беременной, приема противосудорожных и некоторых психотропных препаратов. При этом большинство случаев изолированных ДГ являются спорадическими с низким риском повторения в семье.
В экспериментах на животных показано влияние ретинола на процесс возникновения ВДГ и гипоплазии легких за счет супрессии гена Midkine, участвующего в выработке фактора роста легочной ткани (RA-responsive growth factor) [20–22]. Роль уровня ретинола в развитии ВДГ подтверждена при исследовании новорожденных и их матерей [23]. Также обсуждается влияние диабета беременной, приема противосудорожных и некоторых психотропных препаратов. При этом большинство случаев изолированных ДГ являются спорадическими с низким риском повторения в семье.
Двусторонние изолированные ДГ являются намного более редкой патологией и составляют примерно 1% всех ДГ и более характерны для семейных случаев (не спорадических). Роль генетических причин в возникновении данной аномалии существенно выше. Двусторонние ДГ встречаются в 20% описанных семейных случаев данной патологии [24, 25].
A.P. Machado с соавт. [26] сообщили о случае двусторонней ДГ у двух плодов из монохориальной биамниотической двойни. Высокая конкордантность признака у монозиготных близнецов является убедительным свидетельством наличия одинакового первичного генетического дефекта у обоих плодов.
Представляем наблюдение семейного случая двусторонней изолированной ДГ.
Ребенок пациентки Ш., от вторых своевременных оперативных родов.
Семейный анамнез. Мать: 41 г., соматические заболевания – эпидемический паротит в детстве, анемия. Имеет особенности фенотипа- миопию высокой степени, плоскостопие, гипермобильность суставов кистей рук и стоп.
Брак I, неродственный. Отец: 41 г., здоров (военнослужащий), особенности фенотипа – при гастроскопии обнаружена небольшая ДГ, брак I. Кариотипы супругов 46,ХХ, 46, ХУ. Генеалогия: у матери пациентки – рак молочной железы, у свекра – пупочная грыжа больших размеров (умер от осложнений грыжи).
 I беременность в 1993 г. завершилась своевременными родами, родился мальчик массой 3600 г, ростом 52 см, умер через 2 часа. По данным патолого-анатомического исследования у ребенка имелась ложная левосторонняя ДГ, других аномалий развития не обнаружено. II беременность в 1995 г. завершилась искусственным абортом без осложнений.
I беременность в 1993 г. завершилась своевременными родами, родился мальчик массой 3600 г, ростом 52 см, умер через 2 часа. По данным патолого-анатомического исследования у ребенка имелась ложная левосторонняя ДГ, других аномалий развития не обнаружено. II беременность в 1995 г. завершилась искусственным абортом без осложнений.
III беременность в 2011 г. – настоящая. В I триместре обнаружен кандидозный вульвовагинит, II триместр – без осложнений, биохимический скрининг II – в норме, в сроке 19 недель по данным ультразвукового исследования (УЗИ) у плода обнаружена левосторонняя ДГ больших размеров, единственная артерия пуповины. От молекулярного кариотипирования плода путем кордоцентеза семья отказалась. В III триместре развилось многоводие, анемия.
В 39–40 недель беременности в связи с излитием околоплодных вод без начала родовой деятельности было проведено родоразрешение путем операции кесарева сечения. Извлечен живой доношенный мальчик, массой 3668 г, ростом 51 см. Оценка по шкале Апгар – 2/4/5 баллов, в родильном зале проводились реанимационные мероприятия в полном объеме c достижением стабилизации для проведения транспортировки в профильное отделение.
При поступлении из родильного зала в отделение хирургии, реанимации и интенсивной терапии новорожденных отдела неонатологии и педиатрии крайняя степень тяжести состояния ребенка была обусловлена прогрессированием гипоксемии на фоне легочной гипертензии, ассоциированной с гипоплазией обоих легких. С рождения проводилась высокочастотная осцилляторная искусственная вентиляция легких в максимально жестких режимах, терапия легочной гипертензии ингаляцией оксида азота в дозировке 20–40 ppm, кардиотоническая терапия высокими дозами адреномиметиков (допамин, добутамин, адреналин), вводился перорально силденафил. Диагноз двухсторонней ВДГ подтвержден по данным рентгенологического (рис. 1 см. на вклейке) и ультразвукового обследования. Но, несмотря на проводимую интенсивную терапию, состояние ребенка прогрессивно ухудшалось и через 4,5 часа констатирована смерть ребенка.
Произведена оценка фенотипа ребенка врачом-генетиком, при которой внешних признаков дисэмбриогенеза и признаков синдромальной патологии выявлено не было.
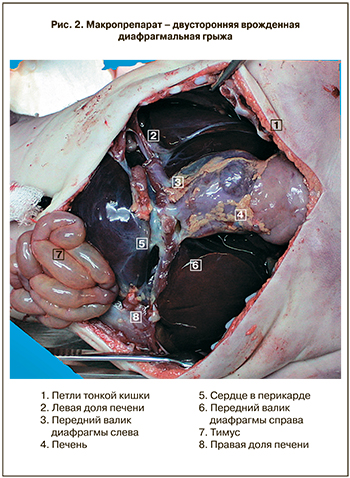
 При патологоанатомическом исследовании тела умершего новорожденного установлено наличие двусторонней ложной ДГ больших размеров. Обнаружены крупные дефекты в правом и левом куполе диафрагмы размерами справа 4х4 см, слева – 5х5 см. Через дефекты в диафрагме часть органов брюшной полости эвентрирована в правую и левую плевральные полости. В правой плевральной полости расположена правая доля печени с желчным пузырём, в левой плевральной полости расположены левая доля печени, селезенка, желудок, часть толстой кишки, в частности, слепая кишка с червеобразным отростком, восходящий и поперечно-ободочный отделы толстой кишки. Правое и левое легкое в состоянии гипоплазии, поджаты к корням легких. Сердце и органы средостения смещены вправо от срединной линии тела (рис. 2 см. на вклейке).
При патологоанатомическом исследовании тела умершего новорожденного установлено наличие двусторонней ложной ДГ больших размеров. Обнаружены крупные дефекты в правом и левом куполе диафрагмы размерами справа 4х4 см, слева – 5х5 см. Через дефекты в диафрагме часть органов брюшной полости эвентрирована в правую и левую плевральные полости. В правой плевральной полости расположена правая доля печени с желчным пузырём, в левой плевральной полости расположены левая доля печени, селезенка, желудок, часть толстой кишки, в частности, слепая кишка с червеобразным отростком, восходящий и поперечно-ободочный отделы толстой кишки. Правое и левое легкое в состоянии гипоплазии, поджаты к корням легких. Сердце и органы средостения смещены вправо от срединной линии тела (рис. 2 см. на вклейке).
Из образцов крови отца, матери и ребенка была выделена ДНК, которая в дальнейшем была проанализирована с помощью метода сравнительной геномной гибридизации на чипах (aCGH). В работе использовали SurePrint G3 180K arrays (Agilent, USA). В качестве референса использовали Male Human Reference DNA (Agilent, USA). Анализ полученных данных показал, что все выявленные CNVs у отца являются доброкачественными, а у матери и ребенка в chr7:26888579-26930268 положении 7-й хромосомы присутствует ранее не описанная CNV размером 41 kB. Преждевременно делать вывод о связи данной CNV с ВДГ, однако в любом случае очевидно, что данная CNV заслуживает дальнейшего исследования.
Семье была проведена медико-генетическая консультация, на которой обсуждались возможные типы наследования ДГ и прогноз будущего потомства.
Обсуждение
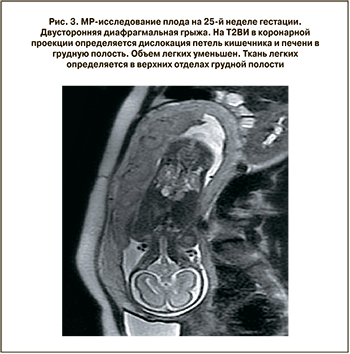
Пренатальная диагностика изолированной двусторонней ДГ является сложной задачей. В нашем наблюдении УЗИ во время беременности позволило предположить наличие только левосторонней ДГ. Данные литературы также свидетельствуют о сложностях ультразвуковой пренатальной диагностики двусторонней ДГ [27, 28]. При данной патологии бóльшую эффективность и высокую информативность в пренатальном периоде показывает магнитно-резонансная томография (МРТ) [27–29], которая, очевидно, должная войти в протокол обследования при наличии ВДГ плода, особенно в случаях предполагаемой хирургической коррекции. Особую трудность при ультразвуковой визуализации представляет дифференцировка ткани печени и легкого. Использование же МРТ позволяет во всех случаях не только достоверно различить ткани различных органов, но и уточнить их топографию, в том числе и при посмертном исследовании [30]. На рис. 3 (см. на вклейке) показаны возможности МРТ в пренатальной диагностике двусторонней ДГ.
Для данной семьи было чрезвычайно важным установить корректный прогноз будущего потомства и рационально определить сценарий планирования следующей беременности.
Тип наследования ВДГ в данном клиническом случае остался неясным, потенциально оказались возможны все распространенные варианты наследования генетической патологии:
- мультифакториальный с семейным накоплением;
- аутосомно-рецессивный – так как имелись два ребенка с однотипной патологией в одном браке;
- аутосомно-доминантный – особенности фенотипов мужа и свекра, умеренно выраженные признаки дисплазии соединительной ткани у пациентки (плоскостопие, гипермобильность суставов кистей рук и стоп, миопия);
- Х-сцепленный рецессивный – два больных сибса были мужского пола. Риск повторного рождения ребенка с ДГ в данной семье определен как высокий, генетический характер патологии не вызывает сомнения.
Учитывая потерю двух детей с ВДГ, а также возраст женщины 41 год, семье было рекомендовано использовать возможности вспомогательных репродуктивных технологий (ВРТ) с применением донорских клеток – и мужских, и женских. Данная тактика позволит свести к минимуму риск рождения ребенка с аномалиями развития, в том числе и с ВДГ. В данной ситуации использование методов ВРТ с полной донацией эмбриона, на наш взгляд, представляется наиболее оптимальным альтернативным способом помощи семье в рождения здорового ребенка.


