

Current progress of genetic and epigenetic studies of cerebral palsy
Cerebral palsy (CP) is a heterogeneous condition, with cases sharing the common feature of non-progressive dysfunction of movement and posture. Although widely studied, the etiology of CP is not yet well understood. It is believed that most cases of CP arise from intrauterine injury to the developing brain. Increasing evidence suggests that (epi)genetic factors might collectively account for a large number of CP cases. In this mini-review, we focus mainly on the genetic and epigenetic aspects of recent progress in understanding the etiology of CP.Chen Shen, Nanbert Zhong
Keywords
mini-review
cerebral palsy
etiology
genetics
epigenetics
Background
Cerebral palsy (CP) is a serious neonatal defect that affects 2–3.5 in 1,000 live births, with an estimated prevalence of 17 million worldwide, and causes great economic and social burdens [1-6]. One Chinese survey reported that the prevalence of cerebral palsy was 2.37 per 1,000 live births in Henan province [3]. CP is heterogeneous in clinical features; although the manifestations vary greatly, cases share the common feature of non-progressive dysfunction of posture and movement control. It can be classified into different neurological subtypes on the basis of the topographic distribution of affected limbs and the predominant quality of the observed motor impairments: spastic diplegia, spastic tetraplegic, spastic hemiplegia, dyskinetic or dystonic, ataxic or hypotonic, or mixed ones. The major subgroup of spastic (diplegic, tetraplegic, or hemiplegia) CP (SCP) accounts for 70–80% of all CP cases [1, 2].
Epidemiologic studies found that more than one third of infants with CP were preterm newborns, and a further 15% of babies had acute encephalopathy after term delivery [7, 8]. Previously, environmental stressors, such as birth asphyxia, were thought to be common factors for developing CP. Currently, however, it is believed that in most cases, CP arises from intrauterine injury to the developing brain, and a few cases of CP are due solely to severe hypoxia or ischemia at birth. Many scientists have recognized that CP usually arises from intrauterine disturbance (prenatally) in the development of the brain, especially between 24 weeks of gestation and birth. Both the genesis and the adverse perinatal clinical conditions could contribute to the etiologies of CP [1, 2]. Some studies suggested that the hypoxia-ischemia or prenatal infection/inflammation effects could lead to a common pathway of perinatal brain injury characterized by neuronal excitotoxicity, apoptosis, and microglial activation during the critical period of development. Meanwhile, increasing evidence indicates that genetic factors might collectively account for a large number of CP cases.
This mini-review aims at carrying out systematic research of the complex CP etiology, especially the heterogeneity underlying the associated or causative genetic variants, by investigating the recent ideas and findings on the pathogenesis of CP.

Association of gene mutation or variation with CP
Genetic studies of familial and sporadic CP cases have been conducted mainly to explore alterations in DNA sequence levels. Most studies of the genetic susceptibilities to CP of common variations, such as single nucleotide polymorphisms (SNPs) and microsatellites, were carried out by association studies using allele-specific microarray/ mass-spectrometry or direct sequencing [9-20]. For instance, Torres-Merino et al. studied whether the risk for CP increases when an expansion of the - 2.5 kb (CCTTT)n microsatellite in the NOS2 gene and an SNP in -C511T of the IL-1β gene promoter occur in individuals with CP after perinatal hypoxic-ischemic encephalopathy [9]. What they found was that the haplotype (CCTTT)14/TT, formed by the expansion of the - 2.5 kb (CCTTT)microsatellite in the NOS2A gene promoter and the -511 C T SNP of the IL-1β gene promoter, could be a useful marker to identify patients who are at high risk for developing CP after hypoxic-ischemic encephalopathy [9]. In a case-control study of 652 patients with CP and 636 healthy controls in a Han Chinese population addressing the susceptibility of genetic variants of NOS1 to CP by using the MassARRAY typing technique, rs10774909, rs3741475, and rs2682826 were found to be associated with neonatal encephalopathy group (CP + NE), whereas SNP rs3782219 was negatively associated with spastic quadriplegia (OR = 0.742) [14]. The polymorphisms rs10431386 (OR = 1.587) and rs7964786 (OR = 1.956) of the MLEC gene have been suggested to function in the pathogenesis of CP in the Chinese population. The C alleles of rs10431386 and rs7964786 were found to inhibit the expression of MLEC in the blood of patients with CP and in the macrophage cell line [15]. In 2015, Kallankari et al. found that the common CCL18 polymorphism rs2015086, together with intraventricular hemorrhage (IVH), had an additive influence on CP susceptibility in a group of children with very low gestational age (VLGA; < 32 weeks) from northern and central Finland (25 cases, 195 controls) [18]. Sun et al. has recently studied a Chinese Han cohort of 763 infants with CP and 738 healthy controls to determine the association of the OLIG2 gene polymorphism with CP [20]. They found a marginal association of the SNP rs6517135 with CP at the genotype level, and the association was greatly strengthened when they focused on the subgroup of infants with CP who suffered from hypoxic-ischemic encephalopathy (HIE) after birth [20].

Investigations based on newly developed sequencing techniques have helped in identifying more de novo gene mutations with the probability of being pathogenic. High-throughput whole-exome sequencing (WES), which is an efficient strategy for finding rare, disease-causing mutations, including de novo mutations, has been applied for CP research [21-25]. In 2013, by homozygosity mapping and exome sequencing, Kruer et al. identified a homozygous c.1100G>A (p.G367D) mutation in ADD3 [22], encoding gamma adducin, in all affected members of a multiplex consanguineous Jordanian family. When examining the functional consequences of the mutation by using patient-derived fibroblasts in vitro, they found that these mutations impaired the normal actin-capping function of adducin, leading to both abnormal proliferation and migration in cultured patient fibroblasts. A novel homozygous AP4M1 mutation c.194_195delAT (p.Y65Ffs*50) was found in two brothers with CP by using WES. These patients showed aggressive behavior in addition to mild dysmorphic features, intellectual disability, spastic paraparesis, and reduced head circumference [23]. McMichael et al. identified and validated 61 de novo protein-altering variants in 43 of 98 (44%) case-parent trios by using WES. Among them, ten de novo mutations in three previously identified disease genes (TUBA1A [n = 2], SCN8A [n = 1], and KDM5C [n = 1]) and in six novel candidate genes (AGAP1, JHDM1D, MAST1, NAA35, RFX2, and WIPI2) were pathogenic for CP potentially, whereas four hemizygous variants on chromosome X in two known disease genes (L1CAM and PAK3) and in two novel candidate genes (CD99L2 and TENM1) were predicted to be pathogenic for CP [24]. In 2015, Schnekenberg et al. identified de novo point mutations of three different genes, KCNC3, ITPR1, and SPTBN2, in patients diagnosed with ataxic CP, by using targeted next-generation sequencing or trio-based exome sequencing [25]. Genetic testing also helps to distinguish CP from other diseases. For instance, benign hereditary chorea (BHC), caused by mutations in the NK2 homeobox 1 gene (NKX2-1), shares clinical features with ataxic and dyskinetic CP, resulting in the possibility of misdiagnosis. In 2013, McMichael et al. found a 7-bp deletion of NKX2-1 gene in a family previously misdiagnosed with ataxic dyskinetic CP [26]. The father in the family and his two children were considered to have ataxic CP until a possible diagnosis of benign familial chorea was made in the children when they were in their early teens. Moreover, individuals with hereditary spastic paraplegia (HSP) with lower limb spasticity as the common clinical feature, especially childhood-onset HSP, could be mistakenly diagnosed with cerebral palsy (CP) [27, 28], although they usually have typical features and specific gene mutations.
Copy number variation (CNV), which is usually referred to as DNA fragment duplication or deletion, has been found to be involved in various neurological diseases, including attention-deficit hyperactivity disorder (ADHD), autism, bipolar disorder, epilepsy, severe intellectual disability, and schizophrenia. CNV has also been found to be associated with CP. Linkage analysis has been used for mapping pathogenic genes in family-based CPs [29]. Applying comparative genome SNP microarray hybridizations, CNVs were also investigated for pathogenic consideration for CP (Table 3). De novo CNVs were identified among 7% of 147 Canadian families, comprising different CP subtypes (including 37.4% with hemiplegia). The identified de novo rare CNVs that consist of genes GRIK2, LAMA1, DMD, PTPRM, and DIP2C may be associated with the brain development. They have a potential impact on the etiology of CP, which were determined to be involved in 18/97 (18.6%) CP cases [30, 31].
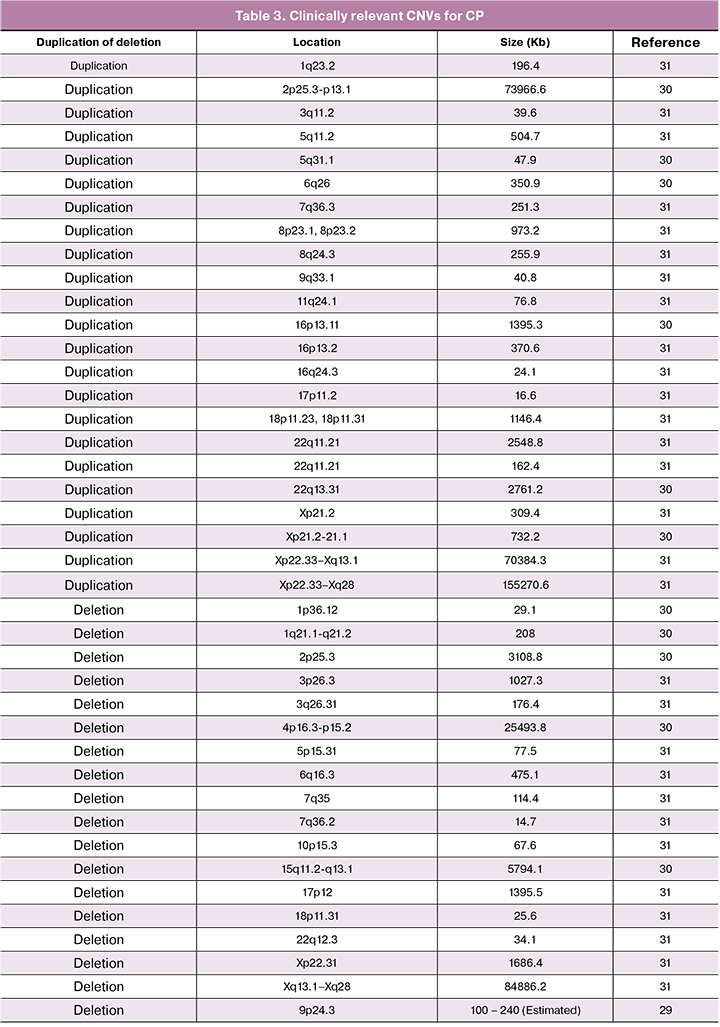
After reviewing the published studies, we summarized the relevant gene loci that are associated with CP in Tables 1-3. Common variations on multiple chromosomes, including 2, 4, 7, 11, 12, 13, 17, 19, and 21, were found to be associated with the onset of CP; point or small-scale gene mutations were identified in the 1, 2, 3, 7, 9, 10, 1, 12, 14, 15, 19, and X chromosomes. CP-relevant CNVs were suggested in nearly all chromosomes.
Epigenetic regulations as the etiology of CP
DNA methylation is one of the most important epigenetic regulation patterns that can be at least partially inherited. The reduced representation bisulfate sequencing in the whole-genome scale was used to identify the DNA methylation profile of CP. Epigenome-wide analysis in newborn blood spots from monozygotic twins discordant for CP revealed consistent regional differences in DNA methylation [32, 33]. In addition, a diagnostic model based on genome-wide methylation profile has been suggested to present both good specificity and good sensitivity for distinguishing young children with spastic CP from non-CP healthy controls [34].
Non-coding RNAs are believed to be important participants in epigenetic regulation. Studies have suggested that microRNAs (miRNAs or miRs) participating in the dysregulation of this process represent the pathogenesis of white matter injury (WMI), which results in cognitive impairment, behavioral disorders, and CP [35]. In 2018, Li et al. discovered a relationship between a functional nucleotide polymorphism in a miRNA coding region and the predisposition of Chinese children to CP [36]. The levels of mature miR-124 were down-regulated by the C-to-T variation in vitro, and the reduction of miR-124 resulting from the C-to-T change led to the less efficient inhibition of the target genes ITGB1, LAMC1, and BECN1, which may play important roles during the development of the nervous system [34].
Studies on gene expression in CP
Genetic and epigenetic alterations can directly alter the expression of the related genes. In addition, complicated regulation mechanisms for genomes must be considered in gene expression. A distant enhancer for the gene of double-sex and mab-3 related transcription factor 3 (DMRT3) was located approximately 120 kb downstream of the cis-regulatory element of the RAR/RXR-binding site within the chromosome region of 9p24.3. Previously it was identified to be deleted and cause congenital neurodegenerative diseases resembling CP wherein congenital hypotonia appeared at first but evolved to spastic quadriplegia with accompanying transient nystagmus over the first year [29]. It was also found that DMRT3 can work as a locomotion coordinator in horses and was highly expressed in the developing mouse forebrain and in some interneurons in the spinal cord, suggesting that the deletion of the cis-regulatory element for DMRT3 in humans may cause impaired development of the forebrain and gait abnormalities, leading to spastic CP [37]. Genome-wide gene transcriptions were investigated in tissues obtained from children with CP [38, 39]. An earlier study found that the transcriptional profiles performed on spastic muscle of CP patients were not characteristic of those observed in other disease states such as Duchenne muscular dystrophy and immobilization-induced muscle atrophy [38]. Another study found that the majority of altered transcripts were related to the increased extracellular matrix expression in CP and a decrease in metabolism and ubiquitin ligase activity, and the increase in extracellular matrix products correlated with disability [39]. In addition, certain studies measured the protein levels mainly for gene variation–based function studies [11-13, 18] or for diagnostic marker screening. Although children with CP exhibited increased energy expenditure during movement, the fiber-type–specific succinate dehydrogenase (SDH) activity, which reflects skeletal muscle mitochondrial oxidative capacity, was similar in children with CP and healthy children without CP [40].
Prenatal exposure as risk factors for CP
One study evaluated the incidence of CP within a population at risk for spontaneous preterm birth that was stratified into subgroups by gestational age (GA) and found that the risk for CP decreased with advancing GA [40]. Apart from the genetic aspect, placental pathology, intrauterine infection, maternal diseases (infection, heart disease, hypertension, anemia, diabetes, and kidney disease) during pregnancy, pregnancy complications, and even environment pollution (air, water, and food) might all influence the onset of CP [3, 41]. Therefore, intrauterine development of fetal brain, with a focus on synaptic function, deserves intensive investigation.
Summary
Recent studies on the risk or causative factors for CP, especially those providing emerging evidence of the genetic aspects, have improved our knowledge of the etiology of CP. However, there is still a long way to go to achieve adequate understanding of CP to reduce its incidence.
References
- Colver A., Fairhurst C., Pharoah P.O. Cerebral palsy. Lancet. 2014; 383(9924): 1240-9.
- Graham H.K., Rosenbaum P., Paneth N., Dan B., Lin J.P., Damiano D.L. et al. Cerebral palsy. Nat. Rev. Dis. Primers. 2016; 2: 15082. https://dx.doi.org/10.1038 / nrdp.2015.82.
- Yuan J., Wang J., Ma J., Zhu D., Zhang Z., Li J. Paediatric cerebral palsy prevalence and high-risk factors in Henan Province, Central China. J. Rehabil. Med. 2019; 51(1): 47-53. https://dx.doi.org/10.2340/16501977-2486.
- Wang F., Cai Q., Shi W., Jiang H., Li N., Ma D. et al. A cross-sectional survey of growth and nutritional status in children with cerebral palsy in west China. Pediatr. Neurol. 2016; 58: 90-7. https://dx.doi.org/10.1016/j.pediatrneurol.2016.01.002.
- Asgarshirazi M., Farokhzadeh-Soltani M., Keihanidost Z., Shariat M. Evaluation of feeding disorders including gastro-esophageal reflux and oropharyngeal dysfunction in children with cerebral palsy. J. Fam. Reprod. Health. 2017; 11(4): 197-201.
- Mei C., Reilly S., Reddihough D., Mensah F., Green J., Morgan P.L. Activities and participation of children with cerebral palsy: Parent perspectives. Disabil. Rehabil. 2015; 37(23): 2164-73.
- McIntyre S., Badawi N., Brown C., Blair E. Population case-control study of cerebral palsy: Neonatal predictors for low-risk term singletons. Pediatrics. 2011; 127: e667-73.
- Mallard C., Davidson J.O., Tan S., Green C.R., Bennet L., Robertson N.J., Gunn A.J. Astrocytes and microglia in acute cerebral injury underlying cerebral palsy associated with preterm birth. Pediatr. Res. 2014; 75(1-2): 234-40.
- Torres-Merino S., Moreno-Sandoval H.N., Thompson-Bonilla M.D.R, Leon J.A.O., Gomez-Conde E., Leon-Chavez B.A. et al. Association between rs3833912/rs16944 SNPs and risk for cerebral palsy in Mexican children. Mol. Neurobiol. 2019; 56(3): 1800-11. https://dx.doi.org/10.1007/s12035-018-1178-6.
- Shang Q., Zhou C., Liu D., Li W., Chen M., Xu Y. et al. Association between osteopontin gene polymorphisms and cerebral palsy in a Chinese population. Neuromolecular Med. 2016; 18(2): 232-8. https://dx.doi.org/10.1007/s12017-016-8397-7.
- Khankhanian P., Baranzini S.E., Johnson B.A., Madireddy L., Nickles D., Croen L.A., Wu Y.W. Sequencing of the IL6 gene in a case-control study of cerebral palsy in children. BMC Med. Genet. 2013; 14: 126. https://dx.doi.org/10.1186/1471-2350-14-126.
- Bi D., Chen M., Zhang X., Wang H., Xia L., Shang Q. et al. The association between sex-related interleukin-6 gene polymorphisms and the risk for cerebral palsy. J. Neuroinflammation. 2014; 11: 100. https://dx.doi.org/10.1186/1742-2094-11-100.
- Hollegaard M.V., Skogstrand K., Thorsen P., Nørgaard-Pedersen B., Hougaard D.M., Grove J. Joint analysis of SNPs and proteins identifies regulatory IL18 gene variations decreasing the chance of spastic cerebral palsy. Hum. Mutat. 2013; 34(1): 143-8. https://dx.doi.org/10.1002/humu.22173.
- Yu T., Xia L., Bi D., Wang Y., Shang Q., Zhu D. et al. Association of NOS1 gene polymorphisms with cerebral palsy in a Han Chinese population: A case-control study. BMC Med. Genomics. 2018; 11(1): 56. https://dx.doi.org/10.1186/s12920-018-0374-6.
- Shi W., Zhu Y., Zhou M., Ruan Y., Chen X., Chen X. Malectin gene polymorphisms promote cerebral palsy via M2-like macrophage polarization. Clin. Genet. 2018; 93(4): 794-9. https://dx.doi.org/10.1111/cge.13149.
- Bi D., Wang H., Shang Q., Xu Y., Wang F., Chen M. et al. Association of COL4A1 gene polymorphisms with cerebral palsy in a Chinese Han population. Clin. Genet. 2016; 90(2): 149-55. https://dx.doi.org/10.1111/cge.12723.
- O’Callaghan M.E., Maclennan A.H., Gibson C.S., McMichael G.L., Haan E.A., Broadbent J.L. et al. Genetic and clinical contributions to cerebral palsy: a multi-variable analysis. J. Paediatr. Child Health. 2013; 49(7): 575-81. https://dx.doi.org/10.1111/jpc.12279.
- Kallankari H., Huusko J.M., Kaukola T., Ojaniemi M., Mahlman M., Marttila R. et al. Cerebral palsy and polymorphism of the chemokine CCL18 in very preterm children. Neonatology. 2015; 108(2): 124-9. https://dx.doi.org/10.1159/000430765.
- Stoknes M., Lien E., Andersen G.L., Bao Y., Blackman J.A., Lie R.T., Vik T. Child apolipoprotein E gene variants and risk of cerebral palsy: Estimation from case-parent triads. Eur. J. Paediatr. Neurol. 2015; 19(3): 286-91. https://dx.doi.org/10.1016/j.ejpn.2014.12.017.
- Sun L., Xia L., Wang M., Zhu D., Wang Y., Bi D. et al. Variants of the OLIG2 gene are associated with cerebral palsy in Chinese Han infants with hypoxic-ischemic encephalopathy. Neuromolecular Med. 2019; 21(1): 75-84. https://dx.doi.org/10.1007/s12017-018-8510-1.
- Tüysüz B., Bilguvar K., Koçer N., Yalçınkaya C., Çağlayan O., Gül E. et al. Autosomal recessive spastic tetraplegia caused by AP4M1 and AP4B1 gene mutation: Expansion of the facial and neuroimaging features. Am. J. Med. Genet. A. 2014; 164A(7): 1677-85. https://dx.doi.org/10.1002/ajmg.a.36514.
- Kruer M.C., Jepperson T., Dutta S., Steiner R.D., Cottenie E., Sanford L. et al. Mutations in γ adducin are associated with inherited cerebral palsy. Ann. Neurol. 2013; 74(6): 805-14.
- Jameel M., Klar J., Tariq M., Moawia A., Altaf Malik N., Seema Waseem S. et al. A novel AP4M1 mutation in autosomal recessive cerebral palsy syndrome and clinical expansion of AP-4 deficiency. BMC Med. Genet. 2014; 15: 133. https://dx.doi.org/10.1186/s12881-014-0133-2.
- McMichael G., Bainbridge M.N., Haan E., Corbett M., Gardner A., Thompson S. et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol. Psychiatry. 2015; 20(2): 176-82.
- Parolin Schnekenberg R., Perkins E.M., Miller J.W., Davies W.I., D’Adamo M.C., Pessia M. et al. De novo point mutations in patients diagnosed with ataxic cerebral palsy. Brain. 2015; 138(Pt 7): 1817-32.
- 26. McMichael G., Haan E., Gardner A., Yap T.Y., Thompson S., Ouvrier R. et al. NKX2-1 mutation in a family diagnosed with ataxic dyskinetic cerebral palsy. Eur. J. Med. Genet. 2013; 56(9): 506-9.
- Rainier S., Sher C., Reish O., Thomas D., Fink J.K. De novo occurrence of novel SPG3A/atlastin mutation presenting as cerebral palsy. Arch. Neurol. 2006; 63(3): 445-7.
- Tessa A., Battini R., Rubegni A., Storti E., Marini C., Galatolo D. et al. Identification of mutations in AP4S1/SPG52 through next generation sequencing in three families. Eur. J. Neurol. 2016; 23(10): 1580-7.
- Lerer I., Sagi M., Meiner V., Cohen T., Zlotogora J., Abeliovich D. Deletion of the ANKRD15 gene at 9p24.3 causes parent-of-origin-dependent inheritance of familial cerebral palsy. Hum. Mol. Genet. 2005; 14(24): 3911-20.
- Oskoui M., Gazzellone M.J., Thiruvahindrapuram B., Zarrei M., Andersen J., Wei J. et al. Clinically relevant copy number variations detected in cerebral palsy. Nat. Commun. 2015; 6: 7949. https://dx.doi.org/10.1038/ncomms8949.
- Zarrei M., Fehlings D.L., Mawjee K., Switzer L., Thiruvahindrapuram B., Walker S. et al. De novo and rare inherited copy-number variations in the hemiplegic form of cerebral palsy. Genet. Med. 2018; 20(2): 172-80. https://dx.doi.org/10.1038/gim.2017.83.
- Mohandas N., Bass-Stringer S., Maksimovic J., Crompton K., Loke Y.J., Walstab J. et al. Epigenome-wide analysis in newborn blood spots from monozygotic twins discordant for cerebral palsy reveals consistent regional differences in DNA methylation. Clin. Epigenetics. 2018; 10: 25. https://dx.doi.org/10.1186/s13148-018-0457-4.
- Jiao Z., Jiang Z., Wang J., Xu H., Zhang Q., Liu S. et al. Whole‑genome scale identification of methylation markers specific for cerebral palsy in monozygotic discordant twins. Mol. Med. Rep. 2017; 16(6): 9423-30.
- Crowgey E.L., Marsh A.G., Robinson K.G., Yeager S.K., Akins R.E. Epigenetic machine learning: utilizing DNA methylation patterns to predict spastic cerebral palsy. BMC Bioinformatics. 2018; 19(1): 225. https://dx.doi.org/10.1186/s12859-018-2224-0.
- Xiao D., Qu Y., Pan L., Li X., Mu D. MicroRNAs participate in the regulation of oligodendrocytes development in white matter injury. Rev. Neurosci. 2018; 29(2): 151-60.
- Li H., Wang X.L., Wu Y.Q., Liu X.M., Li A.M. Correlation of the predisposition of Chinese children to cerebral palsy with nucleotide variation in pri-miR-124 that alters the non-canonical apoptosis pathway. Acta Pharmacol. Sin. 2018; 39(9): 1453-62.
- Kubota N., Yokoyama T., Hoshi N., Suyama M. Identification of a candidate enhancer for DMRT3 involved in spastic cerebral palsy pathogenesis. Biochem. Biophys. Res. Commun. 2018; 496(1): 133-9.
- Smith L.R., Pontén E., Hedström Y., Ward S.R., Chambers H.G., Subramaniam S., Lieber R.L. Novel transcriptional profile in wrist muscles from cerebral palsy patients. BMC Med. Genomics. 2009; 2: 44. https://dx.doi.org/10.1186/1755-8794-2-44.
- Smith L.R., Chambers H.G., Subramaniam S., Lieber R.L. Transcriptional abnormalities of hamstring muscle contractures in children with cerebral palsy. PLoS One. 2012; 7(8): e40686.
- Zogby A.M., Dayanidhi S., Chambers H.G., Schenk S., Lieber R.L. Skeletal muscle fiber-type specific succinate dehydrogenase activity in cerebral palsy. Muscle Nerve. 2017; 55(1): 122-24. https://dx.doi.org/10.1002/mus.25379.
- Smith D.D., Sagaram D., Miller R., Gyamfi-Bannerman C. Risk of cerebral palsy by gestational age among pregnancies at-risk for preterm birth. J. Matern. Fetal Neonatal Med. 2018; 1-5. https://dx.doi.org/10.1080/14767058.2018.1536745.
- Trønnes H., Wilcox A.J., Lie R.T., Markestad T., Moster D. Risk of cerebral palsy in relation to pregnancy disorders and preterm birth: A national cohort study. Dev. Med. Child Neurol. 2014; 56(8): 779-85.
Received 20.12.2018
Accepted 22.02.2019
About the Authors
Chen Shen, PhD, Beijing Pediatric Research Institute, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, 56 Nan-Li-Shi Road, Beijing 10045, China, Tel: +861059616983. E-mail: shenchen1110@126.com.Nanbert Zhong, MD, New York State Institute for Basic Research in Developmental Disabilities, 1050 Forest Hill Road, Staten Island, NY 10314, USA. Tel/Fax: (01) 7184945242/4882. Email: Nanbert.zhong@opwdd.ny.gov
For citation: Chen Shen, Nanbert Zhong. Current progress of genetic and epigenetic studies of cerebral palsy. Akusherstvo i Ginekologiya/Obstetrics and Gynecology. 2019; (6):94-101 (in Russian).
https://dx.doi.org/10.18565/aig.2019.6.94-101
Similar Articles

