
Проблема взаимозаменяемости лекарственных препаратов по своей актуальности стоит на одном из первых мест не только в фармацевтической, но и экономической повестке развития страны. Увеличение удельного веса воспроизведенных лекарственных препаратов в общей структуре потребления лекарственных средств является стратегической задачей, реализация которой приводит к позитивным медико-социальным и экономическим тенденциям в обществе.
Правительство Российской Федерации в целях повышения доступности лекарственных средств уделяет большое внимание увеличению производства воспроизведенных лекарственных препаратов, отвечающих современным требованиям качества, эффективности и безопасности.
Согласно определению Федерального закона № 61-ФЗ «Об обращении лекарственных средств» от 12.04.2010 (ред. от 14.07.2022), воспроизведенный лекарственный препарат – лекарственный препарат для медицинского применения, который имеет эквивалентные референтному лекарственному препарату качественный и количественный составы действующих веществ в эквивалентной лекарственной форме, биоэквивалентность или терапевтическая эквивалентность которого соответствующему референтному лекарственному препарату подтверждена соответствующими исследованиями.
Взаимозаменяемость лекарственных препаратов определяется на основании следующих параметров:
- эквивалентность качественных и количественных характеристик фармацевтических субстанций;
- эквивалентность лекарственной формы;
- эквивалентность или сопоставимость вспомогательных веществ;
- идентичность способа введения и применения;
- отсутствие клинически значимых различий при проведении исследования биоэквивалентности или терапевтической эквивалентности;
- соответствие производителя лекарственного средства требованиям надлежащей производственной практики (Good Manufacturing Practice, GMP).
При этом замена референтного лекарственного препарата на воспроизведенный или одного воспроизведенного препарата другим должна обеспечивать соответствующие качество, эффективность и безопасность.
Интегральным показателем декларируемых качеств воспроизведенных лекарственных препаратов является их биоэквивалентность или терапевтическая эквивалентность в отношении референтного препарата.
Прогестерон не только является важнейшим регулятором нормальной репродуктивной функции в матке, яичниках, молочных железах и мозге, но и играет роль в функционировании нерепродуктивных органов кардиоваскулярной, костной, центральной нервной, иммунной систем, метаболических процессов (обмен воды, электролитов, липидов, углеводов, белков, а также компонентов гемостаза и фибринолиза) [1].
В настоящее время отечественный фармацевтический рынок представлен рядом препаратов прогестерона в различных лекарственных формах, предназначенных для подкожного, внутримышечного, перорального, трансдермального, интравагинального введения.
Препараты прогестерона применяются для лечения прогестерондефицитных состояний у женщин: при угрожающем аборте или для предупреждения привычного аборта, при бесплодии вследствие лютеиновой недостаточности, предменструальном синдроме, нарушениях менструального цикла вследствие нарушения овуляции или ановуляции, фиброзно-кистозной мастопатии, для заместительной гормональной терапии (ЗГТ) в пери- и постменопаузе (в сочетании с эстрогенсодержащими препаратами), ЗГТ в случае дефицита прогестерона при функционирующих (отсутствующих) яичниках (донорство яйцеклеток), для предупреждения преждевременных родов у женщин из группы риска, поддержки лютеиновой фазы во время подготовки к экстракорпоральному оплодотворению и поддержки лютеиновой фазы в спонтанном или индуцированном менструальном цикле, при преждевременной менопаузе [2, 3].
Показаниями к применению препаратов прогестерона в форме геля вагинального являются: поддержание лютеиновой фазы в процессе применения вспомогательных методов репродукции, вторичная аменорея, дисфункциональные маточные кровотечения, обусловленные дефицитом прогестерона, ЗГТ в постменопаузе (в комбинации с эстрогенсодержащими препаратами).
В научной литературе имеются данные о сравнительной эффективности препаратов прогестерона при различных путях введения, в том числе систематический обзор, проведенный Кохрейновским сотрудничеством, который не продемонстрировал существенной разницы между эффективностью препаратов прогестерона при внутримышечном и интравагинальном введении с точки зрения частоты живорождения и частоты продолжающихся беременностей [4, 5].
Интравагинальный путь введения прогестерона имеет следующие преимущества: отсутствие локальной болезненности по сравнению с внутримышечным путем введения, отсутствие системных нежелательных реакций по сравнению с пероральным применением, а также быстрая абсорбция, высокая биодоступность и локальный эффект действия на эндометрий [2, 6–8]. Во многом это связано с транспортировкой прогестерона по анатомически близким к матке кровеносным сосудам, а также с отсутствием первичного прохождения и метаболизации в печени, т. к. кровь от влагалища оттекает в систему нижней полой вены, минуя портальную венозную систему [9, 10].
Фармацевтическая компания АО «ВЕРТЕКС» (Россия, Санкт-Петербург) разработала воспроизведенный препарат «Миражэль» – прогестерон в лекарственной форме гель вагинальный 90 мг/доза.
Препарат «Миражэль» является дженериком по отношению к референтному препарату «Крайнон». Для регистрации препарата «Миражэль» в качестве воспроизведенного препарата необходимо было подтвердить, что системная экспозиция исследуемого лекарственного препарата не превышает таковую лекарственного препарата сравнения, т.е. границы оцененных 90% доверительных интервалов для отношений средних геометрических значений фармакокинетических параметров AUC0-t и Cmax находятся в пределах 80,00–125,00% [11].
Как и референтный препарат, препарат «Миражэль» содержит в своем составе поликарбофил, который обладает свойством биоадгезии и может оставаться на вагинальной стенке в течение 3–4 дней, служа платформой для доставки прогестерона, что позволяет увеличить интравагинальное введение препарата для достижения клинического эффекта – полноценно секреторно измененного эндометрия при сохранении низкой концентрации прогестерона в крови. Более стабильные концентрации прогестерона в системном кровотоке приводят к уменьшению нежелательных реакций [12].
Цель проведенного исследования: оценить биоэквивалентность воспроизведенного препарата прогестерона «Миражэль», гель вагинальный 90 мг/доза (АО «ВЕРТЕКС», Россия), и референтного препарата «Крайнон», гель вагинальный 90 мг/доза («Мерк Сероно Лимитед», Великобритания).
Материалы и методы
Клиническое исследование № 30062016-PgtVERT-001 было проведено в Российской Федерации в соответствии с протоколом клинического исследования, требованиями российского законодательства и ЕАЭС, а также международными правилами проведения клинических исследований (ICH GCP).
В исследование были рандомизированы 42 здоровых женщины-добровольца. Выбор женщин в постменопаузе в качестве субъектов исследования исключал воздействие гормонов, выделяемых функциональными яичниками.
Критерии включения: здоровые женщины в возрасте 40–60 лет (включительно) в периоде естественной постменопаузы с интактной маткой, что подтверждалось отсутствием менструаций в течение как минимум 12 месяцев, значениями уровней фолликулостимулирующего гормона и 17β-эстрадиола в сыворотке крови, находящимися в диапазоне концентраций, соответствующем постменопаузе. Масса тела участниц исследования должна была превышать 45 кг, при этом индекс массы тела (ИМТ) – находиться в пределах 18,5–29,9 кг/м2. Подтвержденный диагноз «здорова» определялся как отсутствие отклонений от нормы, выявленных при подробном медицинском анамнезе, полном врачебном осмотре, включающем измерение артериального давления (АД), частоты сердечных сокращений (ЧСС) и частоты дыхательных движений (ЧДД), температуры тела, электрокардиограмму (ЭКГ) в 12 отведениях и по результатам инструментальных и клинических лабораторных исследований.
Женщины должны были воздерживаться от половых контактов в ходе исследования.
В исследование не включались: женщины, имеющие противопоказания к применению препаратов прогестерона, в том числе с известной гиперчувствительностью, кровянистыми выделениями из половых путей, тромботическими заболеваниями в анамнезе. Также в исследовании не могли участвовать добровольцы с любыми острыми и хроническими заболеваниями, перенесшие гинекологические операции, принимающие или принимавшие гестагены менее чем за 6 месяцев до включения в исследование. Прием лекарственных препаратов, оказывающих выраженное влияние на гемодинамику, функцию печени, также являлся критерием невключения в исследование.
Дизайн исследования
Исследование проведено по открытой рандомизированной перекрестной схеме с 4 периодами, 2 последовательностями введения препаратов с интервалом 14 дней, с участием здоровых добровольцев женского пола. Исследование было открытым, однако сотрудники аналитической лаборатории, проводящие определение концентрации прогестерона в образцах плазмы крови, не имели доступа к схеме рандомизации до окончания статистического анализа данных.
Этап скрининга включал набор добровольцев и проведение их обследования с целью определения критериев включения/невключения.
В начале первого периода участницам исследования присваивались рандомизационные коды. Эти коды определяли последовательность введения исследуемого препарата (T) и препарата сравнения (R) (TRTR или RTRT) в соответствии со схемой рандомизации. Рандомизация была сбалансированной внутри каждой группы. Рандомизационный код каждой участниц исследования не менялся в течение всего периода исследования.
Исследование предполагало 28 визитов для каждой из участниц, общая продолжительность клинического этапа исследования составила около 7 месяцев.
Для оценки биоэквивалентности исследуемый препарат и препарат сравнения вводили добровольцам интравагинально дважды с интервалом 14 дней в дозе 90 мг прогестерона (1,125 г геля исследуемого препарата «Миражэль» (АО «ВЕРТЕКС», Россия) и 1,125 г геля препарата сравнения «Крайнон» («Мерк Сероно Лимитед», Великобритания)). Данная доза не превышала терапевтическую дозу прогестерона. Введение препаратов осуществлялось с использованием специальных одноразовых аппликаторов квалифицированным медицинским персоналом в соответствии с инструкцией по применению. Контроль введения препаратов осуществлялся координатором исследования. Во время проведения исследования никакие другие лекарственные средства не назначались.
Фармакокинетические параметры и статистический анализ данных
На основании полученных фармакокинетических кривых для каждого добровольца были рассчитаны индивидуальные фармакокинетические параметры прогестерона, необходимые для оценки биоэквивалентности сравниваемых препаратов: максимальная концентрация Сmax и площадь под фармакокинетической кривой AUC0-t. Для получения дополнительной информации рассчитывались также площадь под фармакокинетической кривой AUC0-∞, время достижения максимальной концентрации Tmax, среднее время удержания в кровотоке MRT0-t, константа скорости терминальной элиминации kel, период полувыведения T1/2, отношение максимальной концентрации к площади под фармакокинетической кривой Cmax/AUC0-t, отношение AUC0-t/AUC0-∞.
Значения концентрации «ниже предела количественного определения» (BLQ) до введения препаратов в расчетах фармакокинетических параметров и дескриптивной статистики рассматривались как нулевые значения, в последующих точках – как пропущенные значения (missing values).
На каждом этапе исследования образцы крови отбирались в 20 временных точках из локтевой вены добровольцев: до введения (-10, -1, и -0,25 ч) и через 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 18, 24, 36, 48, 60, 72 и 96 ч после введения препарата.
Корректировки значений концентрации прогестерона после введения препаратов на базовый уровень не требовалось, поскольку концентрация прогестерона во всех образцах плазмы крови, отобранных у добровольцев до введения препаратов на I–IV периодах, была ниже предела количественного определения аналитического метода.
Расчет параметров дескриптивной статистики, дисперсионный анализ и оценка биоэквивалентности сравниваемых препаратов проводились с использованием валидированного программного обеспечения Phoenix Winnonlin (Версия 8.0).
Статистический анализ данных по безопасности (расчет параметров дескриптивной статистики, критерий Стьюдента, дисперсионный анализ, критерий Уилкоксона) проводился с использованием SPSS Statistics 20.0.
Для значений концентрации прогестерона и всех фармакокинетических параметров рассчитывались следующие параметры дескриптивной статистики по этапам исследования и по препаратам: среднее арифметическое значение, среднее геометрическое значение, стандартное отклонение среднего результата, максимальное и минимальное значения, размах, медиана, коэффициент вариации.
Дисперсионный анализ (ANOVA) был выполнен на логарифмически преобразованных фармакокинетических параметрах AUC0-t и Cmax прогестерона на уровне значимости α=5%.
Вывод о биоэквивалентности сравниваемых препаратов был сделан с использованием подхода, основанного на оценке 90% доверительных интервалов для отношений средних геометрических значений фармакокинетических параметров AUC0-t и Cmax прогестерона. В соответствии с протоколом исследования сравниваемые препараты считаются биоэквивалентными, если границы оцененных 90% доверительных интервалов для отношений средних геометрических значений фармакокинетических параметров AUC0-t и Cmax находятся в пределах 80,00–125,00%.
Аналитический анализ данных
Отбор образцов крови производился через кубитальные гепаринизированные катетеры или с использованием одноразовых шприцов в вакутейнеры, содержащие гепарин лития в качестве антикоагулянта.
Образцы крови были центрифугированы при 1500 g в течение 10 минут при температуре около +4°C. Центрифугирование проводилось не позднее чем через 15 минут после отбора образца крови. Образцы плазмы крови были заморожены в вертикальном положении и хранились при температуре не выше -20°C.
Концентрация прогестерона в образцах плазмы крови добровольцев определялась методом высокоэффективной жидкостной хроматографии с тандемным масс-спектрометрическим детектированием.
В ходе валидации было показано, что метод является чувствительным, достоверным и селективным в диапазоне концентраций 0,100–20,000 нг/мл. Нижний предел количественного определения аналитического метода составлял 0,100 нг/мл.
В качестве матрицы для приготовления калибровочных стандартов и образцов контроля качества использовалась гепаринизированная плазма крови человека. Стандартный образец прогестерона-d9 использовали в качестве внутреннего стандарта.
Изучение безопасности
Оценка безопасности и регистрация нежелательных явлений проводились на протяжении всего исследования. Наличие нежелательных явлений оценивалось по жалобам добровольцев, а также по данным физикального осмотра, осмотра гинеколога, измерения основных жизненных показателей (АД, ЧСС, ЧДД и температуры тела), результатам ЭКГ в 12 отведениях и результатам анализа лабораторных параметров крови и мочи, определяемых в данном исследовании.
Распределение добровольцев и базовые характеристики
47 добровольцев подписали информированное согласие и участвовали в скрининге. На основе результатов скрининга 44 женщины были включены в исследование. 3 участницы не были включены в исследование, поскольку они не соответствовали требованиям критериев включения и/или у них соблюдался хотя бы один из критериев невключения. Данные 42 участниц, которым были введены исследуемый препарат и препарат сравнения хотя бы по одному разу (добровольцы участвовали в перекрестной схеме исследования), были включены в расчет фармакокинетических параметров, дисперсионный анализ и оценку биоэквивалентности сравниваемых лекарственных препаратов. Схема, представленная на рисунке 1, резюмирует распределение добровольцев в исследовании.

Средний возраст участниц составил 52,79±3,50 года, средний рост – 1,63±0,05 м, масса тела – 70,40±8,79 кг, ИМТ – 26,48±2,97 кг/м2.
Для всех женщин, включенных в исследование, основные жизненные показатели, результаты физикального осмотра, ЭКГ в 12 отведениях, осмотров гинеколога и маммолога, УЗИ органов малого таза, маммограммы и цервикального мазка, а также лабораторных параметров крови и мочи были в пределах нормы на этапе скрининга и соответствовали всем требованиям критериев включения/невключения.
Результаты
Концентрация прогестерона определялась в 3281 образце плазмы крови, полученном от 42 добровольцев (включая образцы досрочно выбывших из исследования добровольцев) в ходе 4 периодов исследования.
Зависимости средних значений концентрации прогестерона в образцах плазмы крови добровольцев по препаратам от времени после введения препаратов представлены на рисунке 2. Различия в индивидуальных фармакокинетических профилях прогестерона после введения сравниваемых препаратов не носили систематического характера. Средние фармакокинетические профили прогестерона после введения исследуемого препарата и препарата сравнения практически не отличались.

Средние значения и стандартные отклонения фармакокинетических параметров прогестерона после введения исследуемого препарата «Миражэль» (АО «ВЕРТЕКС», Россия) и препарата сравнения «Крайнон» («Мерк Сероно Лимитед», Великобритания) суммированы в таблице 1.
Двусторонние 90% доверительные интервалы для отношений соответствующих средних геометрических значений фармакокинетических параметров AUC0-t и Cmax прогестерона представлены в таблице 2.
Коэффициенты внутрииндивидуальной вариабельности для фармакокинетических параметров AUC0-t и Cmax прогестерона, рассчитанные после введения обоих исследуемых препаратов (ISCVtotal) и каждого из исследуемых препаратов отдельно (ISCVWT и ISCVWR), представлены в таблице 3.
Рассчитанные 90% доверительные интервалы для отношений средних геометрических значений фармакокинетических параметров AUC0-t и Cmax прогестерона лежат внутри интервала биоэквивалентности: в соответствии с протоколом исследования сравниваемые препараты считаются биоэквивалентными, если границы оцененных 90% доверительных интервалов для отношений средних геометрических значений фармакокинетических параметров AUC0-t и Cmax находятся в пределах 80,00–125,00%. Для параметра Cmax пределы биоэквивалентности не были расширены, поскольку ISCVWR для Cmax<30%.
Безопасность
В ходе исследования для всех добровольцев популяции безопасности результаты физикального осмотра, осмотра гинеколога, ЭКГ в 12 отведениях и лабораторные параметры мочи оставались в пределах нормы. У ряда добровольцев в ходе исследования наблюдались отклонения от нормы значений основных жизненных показателей, ряда параметров коагулограммы и биохимического анализа крови, которые были зарегистрированы как нежелательные явления.
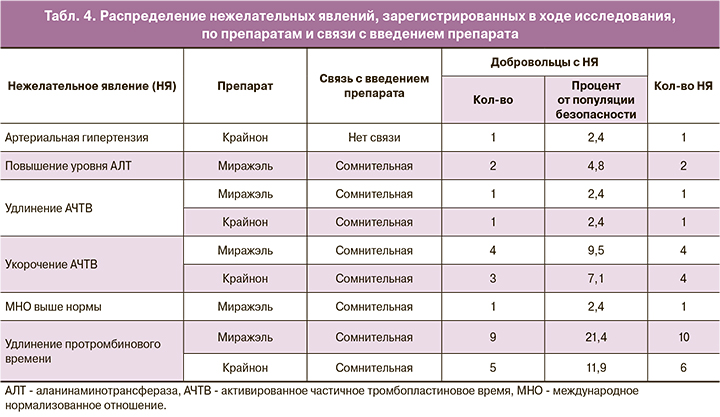
Всего в данном исследовании зарегистрировано 30 нежелательных явлений у 20 добровольцев популяции безопасности (47,6%): 18 нежелательных явлений у 14 добровольцев популяции безопасности (33,3%) после введения исследуемого препарата и 12 нежелательных явлений у 10 добровольцев популяции безопасности (23,8%) после введения препарата сравнения. Все нежелательные явления были несерьезными и легкой степени тяжести.
Все они разрешились без осложнений до завершения исследования, последующего наблюдения не требовали. В случае повышения АД для купирования нежелательного явления потребовалось применение сопутствующей терапии. Из 30 нежелательных явлений 29 имели, по мнению исследователей, сомнительную связь с введением исследуемых препаратов, еще одно нежелательное явление было расценено как не связанное с терапией. Таким образом, в данном исследовании нежелательных явлений, которые были бы достоверно, вероятно или возможно связаны с введением исследуемого препарата или препарата сравнения, не зарегистрировано.
Распределение нежелательных явлений, зарегистрированных в ходе исследования, по препаратам и связи с введением препарата представлено в таблице 4.
На основании полученных данных можно сделать вывод, что исследуемый препарат «Миражэль», гель вагинальный 90 мг/доза (АО «ВЕРТЕКС», Россия), не уступает зарегистрированному препарату сравнения «Крайнон», гель вагинальный 90 мг/доза («Мерк Сероно Лимитед», Великобритания), по таким показателям безопасности, как влияние на основные жизненные показатели, параметры физикального осмотра, осмотра гинеколога, ЭКГ в 12 отведениях, лабораторные параметры крови и мочи, определяемые в данном исследовании, а также частота связанных с введением препарата нежелательных явлений.
Обсуждение
Исследование проводилось с целью подтверждения биоэквивалентности исследуемого препарата таковой референтного препарата.
Предыдущие исследования сообщали о значении Cmax прогестерона 10,51 нг/мл после интравагинального введения препарата «Крайнон» здоровым женщинам в постменопаузе [11]. Это значение сравнимо со значениями, полученными в настоящем исследовании: средние Cmax для препарата «Крайнон» составляли 10,912±3,651 нг/мл, для исследуемого препарата «Миражэль» – 11,497±3,693 нг/мл.
Заключение
Границы оцененных двусторонних 90% доверительных интервалов для отношений средних геометрических значений фармакокинетических параметров AUC0-t и Cmax находятся в пределах 80,00–125,00%. Эти данные подтверждают, что исследуемый препарат «Миражэль», гель вагинальный 90 мг/доза (АО «ВЕРТЕКС», Россия), биоэквивалентен препарату сравнения «Крайнон», гель вагинальный 90 мг/доза («Мерк Сероно Лимитед», Великобритания). Препарат «Миражэль» не уступал по параметрам безопасности препарату «Крайнон».


