

The role of chromosomal abnormalities in fetal congenital heart defects
Pak V.S., Tetruashvili N.K., Bokeriya E.L., Shubina Je,. Zaretskaya N.V., Bolshakova A.S., Lyushnina D.G., Kuznetsova M.V., Mikhailovskaya G.V., Sadelov I.O., Trofimov D.Yu.
Objective: The objective of this study was to investigate the frequency of chromosomal abnormalities in various nosological forms of fetal congenital heart defects (CHDs). Materials and methods: The study included 72 pregnant women who received a prenatal diagnosis of fetal CHD. Between 15 and 30 weeks of pregnancy, the women underwent invasive prenatal diagnostic procedures. Fetal DNA testing was performed in two stages. The first stage was detection of abnormalities in chromosomes 13, 18, 21, X, and Y using polymerase chain reaction analysis of short-tandem repeat (STR) markers (STR-PCR). The second stage was chromosomal microarray analysis (CMA). Results: Among 72 women, congenital heart defects were found in combination with chromosomal abnormalities in 16 сases (22.2% of total cases). Of them, trisomy 21 was identified in 4 cases (5.6%), trisomy 18 in 2 cases (2.8%), microdeletion from chromosome 22 in 8 cases (11.1%), microdeletion from chromosome 12 in 1 case (1.4%), microdeletion from chromosome 1 in 1 case (1.4%). Additionally, aneuploidies were detected in 6 cases (8.3%) using quantitative fluorescent polymerase chain reaction (QF-PCR), and all copy number variations were confirmed by chromosomal microarray analysis (CMA) in 10 cases (13.9%). Conclusion: The study found that fetal congenital heart defects were associated with chromosomal abnormalities in 22% of cases (in 16 cases out of 72). The majority of abnormalities were related to pathogenic gene copy number variants in 62.5% of cases (in 10 cases out of 16). Based on the findings, the preferred invasive method of prenatal diagnostics should be considered to be chromosomal microarray analysis, as it provides comprehensive genetic information.
Authors' contributions: Pak V.S., Tetruashvili N.K., Bokeriya E.L., Trofimov D.Yu. – the concept and design of the study; Pak V.S., Zaretskaya N.V, Bolshakova A.S., Lyushnina D.G., Kuznetsova M.V., Mikhailovskaya G.V., Sadelov I.O. – material collection and processing; Pak V.S., Bolshakova A.S. – statistical data processing; Pak V.S., Tetruashvili N.K. – article writing; Tetruashvili N.K., Bokeriya E.L. Shubina Je., Bolshakova A.S., Sadelov I.O., Trofimov D.Yu. – article editing.
Conflicts of interest: The authors declare that they have no conflicts of interest.
Funding: The study was carried out within the frames of the State Assignment on the topic “Development of test-system for prenatal diagnostics of fetal cardiac pathology”.
Ethical Approval: The study was approved by the Ethics Committee of the National Medical Research Center for Obstetrics, Gynecology and Perinatology named after Acaemician V.I. Kulakov of the Ministry of Health of Russia.
Patient Consent for Publication: All patients provided informed consent for the publication of their data.
Authors' Data Sharing Statement: The data supporting the findings of this study are available on request from the corresponding author after approval from the principal investigator.
For citation: Pak V.S., Tetruashvili N.K., Bokeriya E.L., Shubina Je,. Zaretskaya N.V., Bolshakova A.S., Lyushnina D.G., Kuznetsova M.V., Mikhailovskaya G.V., Sadelov I.O., Trofimov D.Yu. The role of chromosomal abnormalities in fetal congenital heart defects. Akusherstvo i Gynecologia/Obstetrics and Gynecology. 2023; (10): 86-93 (in Russian) https://dx.doi.org/10.18565/aig.2023.220
Keywords
congenital heart defect
chromosomal abnormality
22q11.2 deletion syndrome
chromosomal microarray analysis
Congenital heart disease (CHD) dominates among all congenital anomalies and reaches about 30% in the structure of fetal congenital malformation [1]. The incidence of CHD in the population ranges from 6 to 8 per 1000 live births (0.6–0.8) and 10% of stillbirths [2]. The incidence of neonatal mortality due to congenital heart disease is higher in the early neonatal period [3]. About 20 000 babies are born with CHD every year in the Russian Federation. About a quarter of them require surgical treatment in the first days of life [4].
Congenital heart defects (CHDs) are classified according to the severity, the effect on pulmonary blood flow, and the features of hemodynamic disturbances. There are also physiological and anatomical classifications [5]. A number of authors distinguish between isolated CHD and CHD combined with extracardiac anomalies [6].
A number of studies use the classification of congenital heart disease according to Botto L. et al., based on the phenotype and etiology of CHD: 1 – conotruncal anomalies (interrupted aortic arch, tetralogy of Fallot, transposition of the great arteries (TGA), double outlet right ventricle (DORV), truncus arteriosus (TA), 2 – atrial septal defect (ASD), 3 – anomalous pulmonary venous drainage (APVD), 4 – left ventricular outflow tract obstruction (LVOTO) (hypoplastic left heart syndrome (HLHS), coarctation of the aorta/aortic stenosis), 5 – right ventricular outflow tract obstruction (RVOTO), 6 – septal defects, 7 – heterotaxy (HTX), 8 – single ventricle, 9 – combined defects [7].
Fetal congenital heart disease is of diverse etiology and is not sufficiently studied. According to various authors, in about 20–30% of cases, CHD is caused by genetic factors [8]. To assess the genetic basis of CHD formation, it is most advisable to study the groups of defects that have unfavorable or questionable prognosis in terms of disability and mortality in children.
It is proved that congenital heart defects are associated with following aneuploidies: trisomy of chromosome 18 – 95% [9], chromosome 13 – 60–80% [10], chromosome 21 – 40–50% [11], chromosome 9 – 65–80%, chromosome 8 – 25%, monosomy X– 25–50% [12], Klinefelter syndrome – 50%. The literature describes pathologic gene copy number variants (CNVs) in CHD, such as 20р12 – 85–94%, 22q11.2 – 75–80% [13], 4р – 50–65%, 7q11.23 – 53–85%, 8р – 50–75%, 5р – 30–60%, 11q – 56%, 10р – 50% [5]. In turn, meta-analysis by Wang H. et al. described the total proportion of chromosomal abnormalities in CHD. Thus, aneuploidies, 22q11 deletions and other CNVs in fetuses with congenital heart disease was 23% (95% CI 20–26%), 19% (95% CI 16–22%), 2% (95% CI 2–3%) and 4% (95% CI 3–5%), respectively [14].
Some CHDs are associated with monogenic disorders. Prospective cohort meta-analysis by Mone F. et al. (2021) showed that in fetal CHD additional prognostic value of exome sequencing compared to karyotyping and chromosomal microarray analysis (CMA) was about 12.7% [15].
Given the above, identification of the genetics of congenital heart disease makes it possible to determine further pregnancy management tactics, formulate clear indications for termination of pregnancy, and plan the routing for pregnant woman.
The objective of the study was to investigate the frequency of chromosomal abnormalities in various nosological forms of fetal congenital heart defects (CHDs).
Materials and methods
From 2019 to 2023, 72 patients with isolated congenital heart disease that was diagnosed by ultrasound in the regional perinatal centres, were referred for consilium in the National Medical Research Center for Obstetrics, Gynecology and Perinatology named after Academician V.I. Kulakov of the Ministry of Health of Russia (the Center).
Based on fetal echocardiography, the diagnosis of CHD at 15-30 weeks of pregnancy was verified by the specialists of the Center. In the frames of perinatal consilium, all married couples were consulted by obstetrician-gynecologist, cardiac surgeon and geneticist and received total information on diagnosis, and the prognosis of the disease. The Figure shows the design of the study.
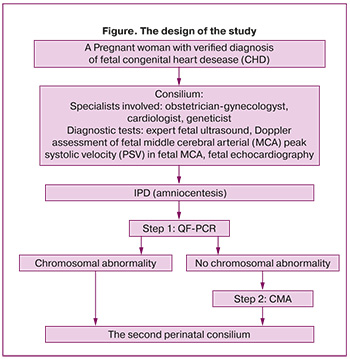
The study included the group of patients with CHD, such as tetralogy of Fallot – 25/72 (34.75%) cases, pulmonary atresia (PA) – 8/72 (11%), interrupted aortic arch/aortic atresia/hypoplastic aortic arch 17/72 (23.6%), transposition of the great arteries (TGA) – 10/72 (13.9%), double outlet right ventricle (DORV) 4/72 (5.6%), common arterial trunk – 4/72 (5.6%), the patent atrioventricular canal – 1/72 (1.4%), hypoplastic left heart syndrome (HLHS) – 1/72 (1,4%), single ventricle – 1/72 (1,4%), agenesis of the venous duct – 1/72 (1,4%) case (Table 1).

Given the diagnosed fetal CHD, to determine the influence of the genetic factor to occurrence of fetal CHD, it was suggested to perform invasive prenatal diagnosis (IPD) at 15–30 weeks of pregnancy. It is noteworthy, that indicators of the screening test in all pregnant women in the first trimester were normal, and due to this fact, the patients did not undergo IPD.
Before invasive intervention, all patients have signed voluntary informed consent including detailed information on possible risks and complications of IPD. The sample of 40 ml amniotic fluid was obtained by transabdominal amniocentesis and transported to the Institute of Reproductive Genetics of the Center for chromosome analysis.
Fetal DNA testing was performed in two steps. The first step was detection of abnormalities in chromosomes 13, 18, 21, X, and Y using quantitative fluorescence-polymerase chain reaction (QF-PCR) of short-tandem repeat (STR) markers (according to Technical Requirements 21.20.23-113-46482062-2021) for rapid determination of the most common aneuploidies, which account for more than 95% of all chromosome abnormalities in newborns [16]. The diagnostic test performance of QF-PCR takes 3 days, while standard karyotyping technique for fetal DNA extraction from amniotic fluid takes about 3 weeks. According to the Russian authors, the appropriateness of performing prenatal karyotyping should be discussed individually, since microchromosomal rearrangements cannot be excluded in normal caryotype, and are 2–3 times more common in cases of fetal congenital anomalies [17]. When the aneuploidies were identified, further diagnostic search was stopped. In the absence of abnormalities in chromosomes 13, 18, 21, X, Y, the second step of testing was performed.
The second step of fetal DNA testing was Oligo-SNP chromosomal microarray analysis (CMA) using GeboScan 3000 and CytoScan Optima microarray (Thermo Fisher Scientific, USA) according to the manufacturer’s protocol. The obtained data were analyzed using software package ChАS (“Сhromosome Аnalysis Suite”). Copy number variations (CNVs) were interpreted as five categories based on ACMG variant classification guidelines [18]. The size, location of gene copy number variations, gene composition, description in databases (Clinvar, OMIM, ORPHANET, DECIPHER, DGV) as well as in literature were assessed. Pathogenic and likely pathogenic gene copy number variations were included in the report; variants of unclear clinical significance were included only if they were highly consistent with the clinical picture.
Statistical analysis
Statistical data processing was performed using Microsoft Excel Tables (USA). Qualitative indicators are shown as absolute and relative values (%), the quantitative indicators are presented as arithmetic mean (M) and standard deviation (SD).
Results and discussion
The clinical and anamnestic data of the patients in the study were analyzed.
The mean age of patients was 32.4 years. The number of women over 35 years was 26/72 (36.1% of women). The average length of pregnancy at the time of CHD detection was 20 weeks (SD 3.2, 15 –30 weeks). In 42/72 (58.3%) cases, the patients were primigravida.
When collecting the anamnesis of patients, no influence of household or occupational hazards was identified; the patients were non-smokers and had no bad habits. Household hazards included microclimatic conditions, radiation, microbiological and toxicochemical characteristics of the patients’ place of residence. Early diagnosis of fetal CHD before 22 weeks’ gestation was in 47/72 (65.2%) cases, after 22 weeks in 25/72 (34.7%) cases.
The clinical and anamnestic data of pregnant women with fetal CHD are shown in Table 2.
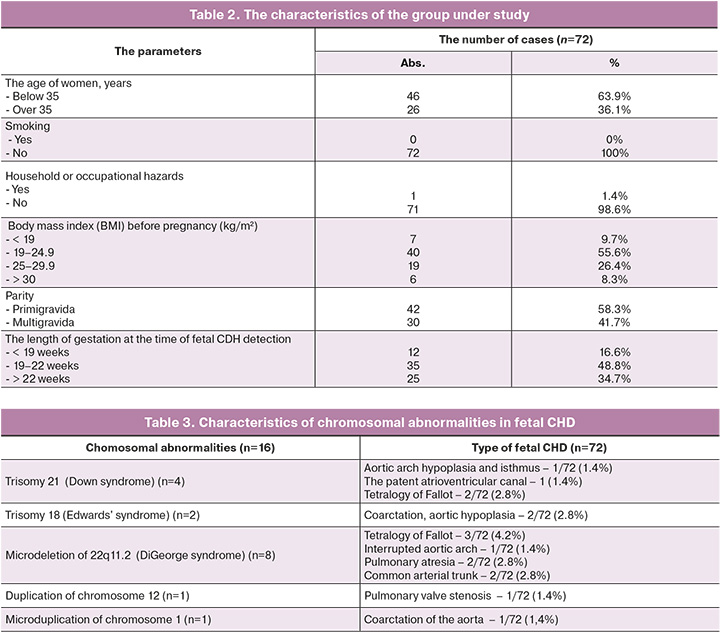
According to the obtained results, CHD associated with chromosomal abnormality was detected in 16/72 (22.2%) cases. Of them, trisomy 21 was identified in 4 cases (5.6%), trisomy 18 in 2 cases (2.8%), microdeletion of 22q11.2 in 8 cases (11.1%), microdeletion of chromosome 12 in 1/72 case (1.4%), microduplication of chromosome 1 in 1/72 case (1.4%). Aneuploidies were detected in 6 cases (8.3%) using quantitative fluorescent polymerase chain reaction (QF-PCR), all copy number variations were detected by CMA in 10 cases (13.9%) (Table 3).
In the cohort of patient in the study, aneuploidies were observed in 8.3% of cases., gene copy number variations in 13.9% of cases, that correlated with the data reported by the American College of Cardiology (aneuploidies in 8 – 10% of cases, gene copy number variations in 25% of cases) [19].
In our study, the incidence rate of Down syndrome was 4/72 (5.5%). Three-fourths of patients refused to continue pregnancy. One family took a decision to continue pregnancy, their born baby died in early neonatal period due to associated neonatal sepsis in combination with hypoplasia of the aortic arch and isthmus. According to the published data, about 50% of patients with Down syndrome have CHD [20]. DYRK1A, RCAN1, DSCAM, COLVI, NRIP1 genes encoded in chromosome 21, are directly involved in occurrence of endocardial cushions [21]. With failure of fusion of endocardial cushions in early embryonic development, congenital heart defects specific for Down syndrome are formed: the patent atrioventricular canal, atrial and interventricular septal defect, and in some cases, tetralogy of Fallot. It is important to note that in our study in case of Down syndrome, the type of fetal CHD heart disease was similar.
In our study, chromosomal abnormality was microdeletion of 22q11.2 in 8/72 (11.1%) cases of fetal CHD; 7/8 patients refused to continue pregnancy; 1 patient took a decision to continue pregnancy and gave birth to full-term baby. At present, the child with tetralogy of Fallot does not require surgical treatment of CHD; further examination and follow-up is undertaken.
The critical region for microdeletion of 22q11.2 includes the TBX1 gene, which plays a key role in formation of 3–4 pharyngeal arches that develop into hard palate, thymus, the conotruncal region of the heart and the great vessels [22]. In embryogenesis of the heart, the right ventricular outflow tract, aorta and pulmonary artery develop from the conotruncal region. When the development of this region of the heart is impaired due to 22q11.2 deletion, congenital heart diseases typical for DiGeorge syndrome occur, such as: interruption of the aortic arch, tetralogy of Fallot, transposition of the great arteries, double outlet right ventricle, common arterial trunk [23]. With 22q11.2 deletion, the type of fetal CHD in our cohort of patients was comparable with the data in world literature [24, 25].
The most common cause of DiGeorge syndrome and a number of other conditions, such as velocardiofacial syndrome, conotruncal anomaly face syndrome, Cayler cardio-facial syndrome is 22q11.2 deletion [26]. DiGeorge syndrome includes conotruncal heart disease, congenital immunodeficiency due [27], hypoparathyroidism (the cause of neonatal seizures due to hypocalcemia), nasopharyngeal pathology, developmental speech disorders cognitive impairment.
Immunodeficiency in children with 22q11.2 deletion is associated with thymic aplasia/hypoplasia, which is responsible for the production of T cells, and leads to frequently recurrent viral and fungal infections. Depending on thymic aplasia or hypoplasia, DiGeorge syndrome is classified as partial or complete [28].
Also, according to Brenner M. et al., the children with CHD in combination with 22q11.2 deletion have worse postoperative outcomes due to defect in the gene encoding glycoprotein Ibβ [29]. It is known that postoperative hemorrhage occurs 2 times more often leading to repeated surgical interventions in the first 24 hours. These complications are associated deficiency of glycoprotein Ibβ, which is necessary for expression of the GPIb-V-IX complex on the surface of platelets, where it functions as the receptor for von Willebrand factor (VWF). The described fact complicates management of this group of patients, who require cardiac surgery, and it should be taken into account when predicting survival at the stage of prenatal family counseling.
According to the data in literature, in 10% of cases 22q11.2 deletion is inherited from one of the parents (inheritance occurs in an autosomal dominant pattern). Given this, for further correct genetic counseling of married couples, it is necessary to offer to undergo CMA according to the trio model (mother-father-fetus) to identify hereditary forms of heart defects [30].
The study by Mustafa H. et al. (2019) showed that in fetal CHD, chromosomal abnormalities are in 36.9% of cases, of them aneuploidies are in 29.5% of cases, and copy number variations in 7.4% [31].
In the Russian Federation, in carrying out IPD in patients with fetal CHD, the first-line testing is standard cytogenic caryotyping. However, Mustafa H. et al. (2019) reported that using CMA in fetal CHD, the diagnostic value increases in 7.4% of cases [31]. According to our study, in 13.9% of cases CHD can be associated with various microdeletion syndromes, and their detection directly depends on diagnostic methods. Our study showed that CMA should be considered to the method of choice in the diagnosis of genetic abnormalities in fetuses with CHD.
According to the obtained data, there were no chromosomal abnormalities in 56/72 (77.7%) of observations. Prolonged pregnancy was in 53/72 (73.6%) case. Antenatal fetal death at 39 weeks of pregnancy was in 1/56 (1.9%) case. Missed miscarriage was at 20 weeks in 1/56 (1.9%) case, neonatal death in early postoperative period was in 2/56 (3.6%) cases.
The study enabled not only to determine the prognosis for survival and health of the fetus/newborn, but also to expand the possibilities of genetic counseling for the family in subsequent pregnancies.
Conclusion
The study showed that there is a category of patients (22%), in whom СHD is associated with chromosomal abnormalities; most part of them relates to pathogenic gene copy number variants (62,5%). The method of choice in carrying out IPD is CMA due to the fact that a number of microdeletion/microduplication syndromes can not be identified by the standard cytogenetic testing.
In our study, the diagnostic value of CMA was 2 times higher bersus QF-PCR.
In carrying out prenatal diagnosis, full information is necessary to identify the genetic causes of fetal congenital heart disease.
Timely information at the prenatal stage about the concomitant fetal chromosomal abnormality gives the opportunity to make a substantiated decision about prolongation or termination of pregnancy. Further research will expand the possibilities of genetic counseling of the families in subsequent pregnancies and predict survival chances of newborns.
References
- Саперова Е.В., Вахлова И.В. Врожденные пороки сердца у детей: распространенность, факторы риска, смертность. Вопросы современной педиатрии. 2017; 16(2): 126-33. [Saperova E.V., Vakhlova I.V. Congenital heart diseases in children: incidence, risk factors, mortality. Current Pediatrics. 2017; 16(2): 126-33. (in Russian)]. https://dx.doi.org/10.15690/vsp.v16i2.1713.
- Taylor K., Elhakeem A., Nader J.L.T., Yang T., Isaevska E., Richiardi L. et al. The effect of maternal pre-/early-pregnancy BMI and pregnancy smoking and alcohol on congenital heart diseases: a parental negative control study. medRxiv. 2020; 2020.09.29.20203786. https://dx.doi.org/10.1101/2020.09.29.20203786.
- Cloete E., Bloomfield F.H., Sadler L., de Laat M.W.M., Finucane A.K., Gentles T.L. Antenatal detection of treatable critical congenital heart disease is associated with lower morbidity and mortality. J. Pediatr. 2019; 204: 66-70. https://dx.doi.org/10.1016/j.jpeds.2018.08.056.
- Бокерия Е.Л. Перинатальная кардиология: настоящее и будущее. Часть I: врожденные пороки сердца. Российский вестник перинатологии и педиатрии. 2019; 64(3): 5-10. [Bokerija E.L. Perinatal cardiology: the present and the future. Part I: congenital heart disease. Russian Bulletin of Perinatology and Pediatrics. 2019; 64(3): 5-10. (in Russian)]. https://dx.doi.org/10.21508/1027-4065-2019-64-3-5-10.
- Министерство здравоохранения Российской Федерации, Союз педиатров России, Ассоциация детских кардиологов России. Федеральные клинические рекомендации по оказанию медицинской помощи детям с врожденными пороками сердца. М.; 2015. [Ministry of Health of the Russian Federation, Union of Pediatricians of Russia, Association of Pediatric Cardiologists of Russia. Federal clinical guidelines for providing medical care to children with congenital heart disease. Moscow; 2015. (in Russian)].
- Qiao F., Wang Y., Zhang C., Zhou R., Wu Y., Wang C. et al. Comprehensive evaluation of genetic variants using chromosomal microarray analysis and exome sequencing in fetuses with congenital heart defect. Ultrasound Obstet. Gynecol. 2021; 58(3): 377-87. https://dx.doi.org/10.1002/uog.23532.
- Botto L.D., Lin A.E., Riehle-Colarusso T., Malik S., Correa A.; National Birth Defects Prevention Study. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res. A Clin. Mol. Teratol. 2007; 79(10): 714-27. https://dx.doi.org/10.1002/bdra.20403.
- Cowan J.R., Ware S.M. Genetics and genetic testing in congenital heart disease. Clin. Perinatol. 2015; 42(2): 373-93, ix. https://dx.doi.org/10.1016/j.clp.2015.02.009.
- Lin A.E., Santoro S., High F.A., Goldenberg P., Gutmark-Little I. Congenital heart defects associated with aneuploidy syndromes: New insights into familiar associations. Am. J. Med. Genet. C. Semin. Med. Genet. 2020; 184(1): 53-63. https://dx.doi.org/10.1002/ajmg.c.31760.
- Yates A.R., Hoffman T.M., Shepherd E., Boettner B., McBride K.L. Pediatric sub-specialist controversies in the treatment of congenital heart disease in trisomy 13 or 18. J. Genet. Couns. 2011; 20(5): 495-509. https://dx.doi.org/10.1007/s10897-011-9373-x.
- Dimopoulos K., Constantine A., Clift P., Condliffe R., Moledina S., Jansen K. et al. Cardiovascular complications of down syndrome: scoping review and expert consensus. Circulation. 2023; 147(5): 425-41. https://dx.doi.org/10.1161/CIRCULATIONAHA.122.059706.
- Huang A.C., Olson S.B., Maslen C.L. A review of recent developments in turner syndrome research. J. Cardiovasc. Dev. Dis. 2021; 8(11): 138. https://dx.doi.org/10.3390/jcdd8110138.
- Lee M.Y., Won H.S., Baek J.W., Cho J.H., Shim J.Y., Lee P.R. et al. Variety of prenatally diagnosed congenital heart disease in 22q11.2 deletion syndrome. Obstet. Gynecol. Sci. 2014; 57(1): 11-6. https://dx.doi.org/10.5468/ogs.2014.57.1.11.
- Wang H., Lin X., Lyu G., He S., Dong B., Yang Y. Chromosomal abnormalities in fetuses with congenital heart disease: a meta-analysis. Arch. Gynecol. Obstet. 2023; 308(3): 797-811. https://dx.doi.org/10.1007/s00404-023-06910-3.
- Mone F., Eberhardt R.Y., Morris R.K., Hurles M.E., McMullan D.J., Maher E.R. et al. Congenital heart disease and the Diagnostic yield with Exome sequencing (CODE) study: prospective cohort study and systematic review. Ultrasound Obstet. Gynecol. 2021; 57(1) :43-51. https://dx.doi.org/10.1002/uog.22072.
- Баранов В.С., Кузнецова Т.В. Новые возможности генетической пренатальной диагностики. Журнал акушерства и женских болезней. 2015; 64(2): 4-12. [Baranov V.S., Kuznetzova T.V. Novel options in Prenatal Genetic Diagnostic. Journal of Obstetrics and Women's Diseases. 2015; 64(2): 4-12. (in Russian)]. https://dx.doi.org/10.17816|/JOWD6424-12.
- Нагорнева С.В., Прохорова В.С., Шелаева Е.В., Худовекова А.М. Анализ частоты выявления врожденных пороков развития у плодов за последние 5 лет (2013-2017). Журнал акушерства и женских болезней. 2018; 67(3): 44-8. [Nagorneva S.V., Prokhorova V.S., Shelaeva E.V., Khudovecova A.M. The prevalence of congenital fetal anomalies for the past 5 years (2013-2017). Journal of Obstetrics and Women's Diseases. 2018; 67(3): 44-8. (in Russian)]. https://dx.doi.org/10.17816/JOWD67344-48.
- https://acmgen.org/wp-content/uploads/2019/11/Tech.
- Pierpont M.E., Brueckner M., Chung W.K., Garg V., Lacro R.V., McGuire A.L. et al. Genetic basis for congenital heart disease: Revisited: a scientific statement from the American Heart Association. Circulation. 2018; 138(21): e653-711. https://dx.doi.org/10.1161/CIR.0000000000000606.
- Asim A., Agarwal S. Congenital heart defects among Down’s syndrome cases: an updated review from basic research to an emerging diagnostics technology and genetic counselling. J. Genet. 2021; 100: 45.
- Mollo N., Scognamiglio R., Conti A., Paladino S., Nitsch L., Izzo A. Genetics and molecular basis of congenital heart defects in Down syndrome: role of extracellular matrix regulation. Int. J. Mol. Sci. 2023; 24(3): 2918. https://dx.doi.org/10.3390/ijms24032918.
- Kloesel B., DiNardo J.A., Body S.C. Cardiac embryology and molecular mechanisms of congenital heart disease: a primer for anesthesiologists. Anesth. Analg. 2016; 123(3): 551-69. https:/dx./doi.org/10.1213/ANE.0000000000001451.
- Ярыгина Т.А., Гасанова Р.М., Большакова А.С., Марзоева О.В., Сыпченко Е.В., Гус А.И. Кардиальная патология в случаях монозиготных двоен с синдромом делеции хромоcомы 22 (22q11DS). Акушерство и гинекология. 2022; 6: 140-51. [Yarygina T.A., Gasanova R.M., Bolshakova A.S., Marzoeva O.V., Sypchenko E.V., Gus A.I. Cardiac pathology in cases of monozygotic twins with chromosome 22 deletion syndrome (22q11DS). Obstetrics and Gynecology. 2022; (6): 140-51. (in Russian)]. https://dx.doi.org/10.18565/aig.2022.6.140-151.
- Goldmuntz E. 22q11.2 deletion syndrome and congenital heart disease. Am. J. Med. Genet. C. Semin. Med. Genet. 2020; 184(1):64-72. https://doi.org/10.1002/ajmg.c.31774.
- Винокурова Е.А., Скрябин Е.Г., Белов В.П. Возможности современной пренатальной диагностики микроделеционного синдрома 22q11.2 (синдрома Ди Джорджи). Российский вестник акушера-гинеколога. 2022; 22(4):39-46. [Vinokurova E.A., Skryabin E.G., Belov V.P. Possibilities of modern prenatal diagnosis of microdeletion syndrome 22q11.2 (DiGeorge syndrome). Russian Bulletin of Obstetrician-Gynecologist. 2022;22(4):39-46. (in Russian)]. https://doi.org/10.17116/rosakush20222204139.
- Óskarsdóttir S., Boot E., Crowley T.B., Loo J.C.Y., Arganbright J.M., Armando M. et al. Updated clinical practice recommendations for managing children with 22q11.2 deletion syndrome. Genet. Med. 2023; 25(3):100338. https://doi.org/10.1016/j.gim.2022.11.006.
- Biggs S.E., Gilchrist B., May K.R. Chromosome 22q11.2 deletion (DiGeorge Syndrome): immunologic features, diagnosis, and management. Curr. Allergy Asthma Rep. 2023; 23(4):213-22. https://doi.org/10.1007/s11882-023-01071-4.
- Boyarchuk O., Volyanska L., Dmytrash L. Clinical variability of chromosome 22q11.2 deletion syndrome. Cent. Eur. J. Immunol. 2017; 42(4):412-7. https://doi.org/10.5114/ceji.2017.72818.
- Brenner M.K., Clarke S., Mahnke D.K., Simpson P., Bercovitz R.S., Tomita-Mitchell A. et al. Effect of 22q11.2 deletion on bleeding and transfusion utilization in children with congenital heart disease undergoing cardiac surgery. Pediatr. Res. 2016; 79(2):318-24. https://doi.org/10.1038/pr.2015.216.
- Maggadottir S.M., Sullivan K.E. The diverse clinical features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome). J. Allergy Clin. Immunol. Pract. 2013; 1(6):589-94. https://doi.org/10.1016/j.jaip.2013.08.003.
- Mustafa H.J., Jacobs K.M., Tessier K.M., Narasimhan S.L., Tofte A.N., McCarter A.R. et al. Chromosomal microarray analysis in the investigation of prenatally diagnosed congenital heart disease. Am. J. Obstet. Gynecol. MFM. 2020; 2(1):100078. https://doi.org/10.1016/j.ajogmf.2019.100078.
Received 14.09.2023
Accepted 20.10.2023
About the Authors
Viktoriia S. Pak, postgraduate, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(913)897-28-49, v_pak@oparina4.ru, https://orcid.org/0009-0002-1444-9071, 4 Acad. Oparin str., Moscow, Russia, 117997.Nana K. Tetruashvili, PhD, Head of the Obstetric Department of Pregnancy Pathology No. 2, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-14-77, n_tetruashvili@oparina4.ru, https://orcid.org/0000-0002-9201-2281,
4 Acad. Oparin str., Moscow, Russia, 117997.
Ekaterina L. Bokeriya, PhD, Researcher at the Department of Patology of Newborn and Prematurely-Born Children No. 2, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-27-05, e_bokeriya@oparina4.ru,
https://orcid.org/0000-0002-8898-9612, 4 Acad. Oparin str., Moscow, Russia, 117997.
Jekaterina Shubina, PhD (Bio), Head of the Laboratory of Genomic Data Analysis, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)531-44-44, e_shubina@oparina4.ru, https://orcid.org/0000-0003-4383-7428,
4 Acad. Oparin str., Moscow, Russia, 117997.
Nadezhda V. Zaretskaya, PhD, Head of the Laboratory of Clinical Genetics of the Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-24-11, n_zaretskaya@oparina4.ru, https://orcid.org/0000-0001-6754-3833,
4 Acad. Oparin str., Moscow, Russia, 117997.
Anna S. Bolshakova, geneticist, Department of Clinical Genetics of the Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-24-11, a_bolshakova@oparina4.ru, https://orcid.org/0000-0002-7508-0899,
4 Acad. Oparin str., Moscow, Russia, 117997.
Daria G. Lyushnina, postgraduate, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(906)308-60-78, d_lyushnina@oparina4.ru, https://orcid.org/0009-0004-3160-8737, 4 Acad. Oparin str., Moscow, Russia, 117997.
Maria V. Kuznetsova, PhD (Bio), Senior Researcher at the Laboratory of Molecular and Genetic Methods of the Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-13-41, mkarja@mail.ru,
https://orcid.org/0000-0003-3790-0427, 4 Acad. Oparin str., Moscow, Russia, 117997.
Galina V. Mikhailovskaya, biologist at the Laboratory of Molecular and Genetic Methods of the Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-13-41, g_mikhailovskaia@oparina4.ru,
4 Acad. Oparin str., Moscow, Russia, 117997.
Igor O. Sadelov, geneticist, Laboratory of Genomic Data Analysis, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-24-10, a_sadelov@oparina4.ru, https://orcid.org/0000-0002-5144-6307, 4 Acad. Oparin str., Moscow, Russia, 117997.
Dmitriy Yu. Trofimov, Dr. Bio. Sci., Professor of the RAS, Corresponding Member of the RAS, Director of the Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-49-51, d_trofimov@oparina4.ru,
https://orcid.org/0000-0002-1569-8486, 4 Acad. Oparin str., Moscow, Russia, 117997.
Similar Articles

