

Expanded genetic testing of a pregnant woman with a congenital heart defect in her fetus
Pak V.S., Tetruashvili N.K., Bokerija E.L., Shubina Je., Zaretskaya N.V., Bolshakova A.S., Maslennikov D.N., Kochetkova T.O., Lyushnina D.G., Trofimov D.Yu.
Relevance: The prevalence of fetal heart defects varies by latitude, ranging from 4 to 50 cases per 1,000 live births. Annually, approximately 283.1 thousand children are born in the Russian Federation with congenital developmental anomalies, and approximately 30% of this population is affected by congenital heart defects (Rosstat data for 2023). In the context of antenatal fetal mortality in the Russian Federation, congenital developmental anomalies rank second, with fetal heart defects being the most common, accounting for 1.2% of stillbirths, and 16.7% of all fetal developmental anomalies.
At the V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, an algorithm for the antenatal examination of pregnant women with fetal heart defects has been developed. This algorithm includes invasive prenatal diagnostics, expanded genetic testing, and genetic counseling for couples.
Objective: To describe the results of examinations of pregnant women with fetal heart defects according to an algorithm developed and implemented by V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia.
Materials and methods: A pregnant woman with a fetal heart defect (coarctation of the aorta and atrial septal defect) underwent invasive antenatal diagnostics and expanded genetic testing. In the first stage, molecular karyotyping was performed using chromosomal microarray analysis of amniotic fluid, followed by sequencing of fetal and parental exomes.
Results: Extensive genetic analysis identified a potentially pathogenic variant within the NR2F2 gene in a fetus with a heart defect. This gene variant is associated with a spectrum of cardiac developmental defects, placental pathology, fetal growth restriction, and sexual developmental disorders. Subsequent Sanger family analysis confirmed that the identified NR2F2 gene variant in the fetus was de novo and not inherited from the parents, indicating a low risk of recurrence in future offspring.
Conclusion: Advanced ultrasound and genetic diagnostic modalities enable the accurate determination of the type of defect, genetic etiology (chromosomal or monogenic), and inheritance pattern during the antenatal period. Additionally, expanded prenatal counseling provides families with comprehensive information about the potential risks, complications, prognosis for disability, and survival of children with heart defects.
Authors’ contributions: Pak V.S., Tetruashvili N.K., Bokerija E.L., Trofimov D.Yu. – conception and design of the study; Pak V.S., Zaretskaya N.V., Bolshakova A.S., Lyushnina D.G., Maslennikov D.N., Kochetkova T.O. – data collection and processing; Pak V.S., Tetruashvili N.K. – drafting of the manuscript; Tetruashvili N.K., Bokerija E.L., Shubina Je., Bolshakova A.S., Trofimov D.Yu. – editing of the manuscript.
Conflicts of interest: The authors have no conflicts of interest to declare.
Funding: The state order under the topic: "Development of a test system for prenatal diagnostics of fetal cardiopathology".
Ethical Approval: The study was reviewed and approved by the Research Ethics Committee of the V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia.
Patient Consent for Publication: The couple signed a voluntary informed consent for the publication of their data.
Authors' Data Sharing Statement: The data supporting the findings of this study are available upon request from the corresponding author after approval from the principal investigator.
For citation: Pak V.S., Tetruashvili N.K., Bokerija E.L., Shubina Je., Zaretskaya N.V., Bolshakova A.S., Maslennikov D.N., Kochetkova T.O., Lyushnina D.G., Trofimov D.Yu.
Expanded genetic testing of a pregnant woman with a congenital heart defect in her fetus.
Akusherstvo i Ginekologiya/Obstetrics and Gynecology. 2024; (11): 83-89 (in Russian)
https://dx.doi.org/10.18565/aig.2024.189
Keywords
congenital heart defect
NR2F2 gene
chromosomal microarray analysis
exome sequencing of congenital heart defect of the fetus
Congenital heart disease (CHD) is a birth defect of the heart that results from abnormal development of the heart and great vessels during pregnancy [1]. Embryonic development of the heart occurs during the 3rd to 9th week of intrauterine development; exposure to unfavorable factors during this period can disrupt the formation of the heart and blood vessels in the fetus [2].
Heart defects (HD) are the most common congenital fetal defects, with a prevalence that varies by latitude, ranging from 4 to 50 cases per 1,000 live births [3]. Annually, approximately 283.1 thousand children are born in the Russian Federation with congenital anomalies (Rosstat data for 2023) [4], and approximately 30% of this population is affected by congenital heart defects [5]. In the context of antenatal fetal mortality in the Russian Federation, congenital anomalies rank second, with fetal heart defects being the most common, accounting for 1.2% of stillbirths and 16.7% of all congenital fetal anomalies [6].
Studies by van Velzen C.L. et al. (2016) [7] have shown that obstetric complications such as premature birth and antenatal fetal death are more common in fetuses with HD than in those without. Furthermore, foreign authors have noted that the presence of isolated fetal HD associated with genetic pathology and valvular regurgitation increases the risk of antenatal loss [8].
Modern ultrasound and genetic research methods enable determination of the type of defect, its genetic (chromosomal or monogenic) cause, and the type of inheritance at the antenatal stage. As part of expanded antenatal counseling, families can receive comprehensive information regarding the possible risks, complications, and prognoses for disability and survival of children with CHD.
Given this information, the management of pregnant women with fetal HD should be comprehensive, including early diagnosis of fetal HD, expanded genetic testing, assessment of the risk of obstetric complications, careful dynamic monitoring of the condition of both the pregnant woman and the fetus, and timely referral to hospital.
Since 2021, the perinatal consultation at the Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of the Russian Federation (V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia), has incorporated expanded genetic studies into the algorithm for examining patients with fetal malformations. This allows the identification of chromosomal and monogenic anomalies in fetuses with HD. A comprehensive approach to managing patients with CHD aids in developing pregnancy management strategies and assessing the prognoses for each couple at the antenatal stage.
The clinical observation presented below illustrates the algorithm for examining and managing pregnant women with fetal HD during the antenatal stage.
This study aimed to describe the results of examinations of pregnant women with fetal heart defects according to an algorithm developed and implemented by the V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia.
The novelty of this diagnostic approach lies in invasive prenatal testing and the implementation of not only chromosomal analysis, but also whole-exome sequencing of fetuses with HD, allowing for the diagnosis of monogenic diseases at the antenatal stage.
Clinical observation
The patient G., is a 28-year-old woman who visited the perinatal consultation at V.I. Kulakov NMRC for OG&P of the Ministry of Health of Russia (Center) at 19–20 weeks of gestation. A fetal HD was detected at her place of residence.
The patient's medical history was significant for chronic pyelonephritis diagnosed at the age of 10 years, no other diseases were reported. Her gynecological history was complicated by the presence of a small uterine fibroid. Her husband had no known chronic conditions.
This was the first spontaneous pregnancy of the patient. The first trimester course was uneventful. During expanded combined prenatal screening at 12 weeks, a single umbilical artery was identified, and the estimated risk of chromosomal pathologies was low. Further management of the pregnancy was continued at her place of residence.
The patient first contacted A.N. Bakulev NMRC of Cardiovascular Surgery in the second trimester (at 19 weeks) after ultrasound examination, during which specialists confirmed a HD: coarctation of the aorta and atrial septal defect (ASD). For further counseling and development of management strategies for the pregnancy, she was referred to a perinatal consultation at the V.I. Kulakov NMRC for OG&P of the Ministry of Health of Russia.
According to a study conducted in 2023 at V.I. Kulakov NMRC for OG&P, the frequency of chromosomal abnormalities in fetal HDs is about 22% [9]. During the perinatal consultation, the patient was offered an expanded genetic study and invasive prenatal diagnosis (amniocentesis). We developed an algorithm for the examination of patients with fetal HDs, which included evaluation of the genetic profile (Figure). In the first stage of genetic examination, chromosomal microarray analysis was performed to exclude the chromosomal pathology of the fetus. As no chromosomal pathology was found, fetal exome sequencing was performed as the second step.
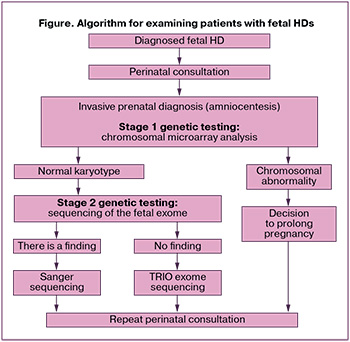
Since there were no contraindications to the invasive procedure at the 19–20 weeks of pregnancy, the patient underwent diagnostic amniocentesis at V.I. Kulakov NMRC for OG&P without complications, during which 45 ml of amniotic fluid was collected.
To exclude the chromosomal pathology of the fetus with a HD, chromosomal microarray analysis was performed, which did not reveal any abnormalities in the fetal karyotype. The family was then subjected to further genetic testing, including fetal exome sequencing. Repeated invasive prenatal diagnostics were not required, as exome sequencing can be performed on residual fetal DNA after chromosomal analysis.
Exome sequencing of the fetus with a HD revealed a previously undescribed heterozygous variant in NR2F2 (15-96337540-G-GT), leading to a frameshift and impaired synthesis of the full-length protein (p.Lys389Ter, NM_021005) at the antenatal stage of examination. According to the existing literature, this variant is associated with various HDs and disorders of sex development (OMIM: 615779, congenital HDs, multiple types, 4; 618901, 46,XX sex reversal 5). A search of the Online Mendelian Inheritance in Man (OMIM) database identified a probable pathogenic variant in the NR2F2 gene on chromosome 15q26 observed in cases with multiple types of HDs, including ventricular septal defect (VSD), atrioventricular septal defect (AVS), double outlet right ventricle, tetralogy of Fallot, hypoplastic left heart syndrome, aortic stenosis, and coarctation of the aorta [10]. Fetal exome sequencing was completed within a five-week period. A heterozygous variant in the NR2F2 gene with an autosomal dominant inheritance pattern was detected. To analyze the origin of this variant, the parents underwent Sanger sequencing. Following a comprehensive examination, it was determined that the parents were not carriers of this variant, and it was assessed as de novo.
After receiving the results of an expanded genetic study at 32 weeks of pregnancy, the couple returned for repeat perinatal consultation at the Center. The patient underwent a second fetal echocardiography during which the functional parameters of the fetus were assessed. The results indicated the presence of multiple HDs, including aortic coarctation, severe hypoplasia of the aortic valve, ascending aorta, and aortic arch, and hypoplasia of the left ventricle, mitral valve, VSD, and hypertrophy of the right ventricle. Additionally, there was evidence of a hydropericardium. Ultrasound diagnostics revealed a low-birth-weight fetus, while uteroplacental and fetoplacental blood flow was normal. During the initial perinatal consultation, the couple was informed of all possible complications and risks to the child after birth and during cardiac surgery for the HD, considering the ultrasound and genetic examination of the fetus. After receiving this information, the couple decided to continue with the pregnancy. A further plan for pregnancy management was developed during the perinatal consultation, including standard examinations according to Order 1130N of the Ministry of Health of the Russian Federation, dated October 20, 2020, a visit to a level 3 hospital to determine the timing of delivery, and planned hospitalization. The patient was to be admitted to a level 3 hospital, with the possibility of transporting the newborn to a cardiac surgery hospital within the first few hours of life.
At 37 weeks of gestation, ultrasound diagnostics and fetal biometric parameters indicated insufficient fetal growth, with an estimated fetal body weight of the 6th percentile.
At 38 weeks of gestation, the patient noted a decline in fetal movement and was immediately admitted to the Center's hospital for further evaluation. Ultrasound examination revealed fetal death. The patient gave birth within 24 h to a stillborn female child weighing 2,775 g and measuring 49 cm in length.
A postmortem examination revealed multiple fetal HDs, including severe hypoplasia of the aortic valve, ascending aorta, and aortic arch; hypoplasia of the left atrial appendage; VSD; right ventricular hypertrophy; signs of fetal hypoxia; and hemorrhage beneath the visceral pleura and under the epicardium. Examination of the placenta showed that it was small for gestational age, with vascular branching, one false umbilical cord knot, three varicose nodes, severe varicose veins, and a single umbilical artery.
Chromosomal microarray analysis was performed using the GenoScan 3000 system on CytoScan Optima microarrays (Thermo Fisher Scientific, USA) in accordance with the manufacturer's instructions. The obtained data were analyzed using the ChAS (Chromosomal Analysis Suite) program. Genetic databases and the existing literature were used to evaluate the examination results.
Fetal DNA sequencing was conducted on a NovaSeq 6000 sequencer, utilizing either the IDT XGen Exome Hyb Panel v2 or the Illumina Exome 2.0 Plus enrichment kit for target fragment enrichment.
The data acquired after sequencing were processed using an algorithm that included alignment of reads to the hg38 reference genome sequence, as well as calling and filtering of variants by quality. Subsequently, the obtained variants were analyzed, annotated, and prioritized using the Ensembl Variant Effect Predictor (VEP), along with an assessment of the significance of variants using SIFT, PolyPhen-2, and SpliceAI.
The population frequencies of the variants were assessed using the Genome Aggregation Database (gnomAD). The OMIM, ClinVar, and LOVD databases were used to assess the clinical significance of identified variants.
Discussion
The etiology of CHD is multifactorial. Environmental factors undoubtedly affect early embryogenesis of the heart and can lead to fetal HDs. These factors include maternal obesity, parental nicotine and alcohol abuse, the use of teratogenic drugs before and during pregnancy, viral infections in the first trimester, and vitamin deficiencies [11, 12].
However, the majority of fetal cases of CHD, between 25 and 40 percent, have a genetic cause [13-15].
The identified heterozygous variant of the nucleotide sequence in the NR2F2 gene (15-96337540-G-GT) results in a frameshift and disrupts the synthesis of the full-length protein (p.Lys389Ter, NM_021005). This gene encodes a transcription factor belonging to the family of steroid and thyroid nuclear receptors. NR2F2 is involved in processes such as cellular differentiation, neurogenesis, organogenesis, fertility, metabolism, and inflammation [16]. A decrease in NR2F2 expression in endothelial cells of the coronary vessels, aorta, and atria leads to inflammation, tissue fibrosis, proliferation, hyperpigmentation, resistance to apoptosis, and an increase in reactive oxygen species [17, 18]. It is important to note that even in cases of successful cardiac surgery for HDs, the risk of an unfavorable prognosis in children with a mutation in this gene increases due to systemic disorders in the vascular and cardiac endothelium.
Furthermore, the literature describes additional clinical manifestations associated with a pathogenic variant of the NR2F2 gene beyond CHD. These include congenital diaphragmatic hernia, fetal growth retardation, hypotension, feeding disorders, congenital and acquired microcephaly, dysmorphic facial features, renal failure, hearing loss, strabismus, and asplenia [19]. Additionally, global literature documents cases of virilization of the external genitalia in girls and sex inversion in boys with a 46XX karyotype [20].
In our clinical observation, insufficient fetal growth was diagnosed at the antenatal stage, with fetal weight, according to fetal biometry findings, in the 6th percentile. Given the monogenic disease of the fetus, we conducted Sanger sequencing of the family (mother, father, and fetus) to determine the significance and risk of inheriting the probable pathogenic variant in the NR2F2 gene. It was established that the parents were not carriers of this variant, allowing us to classify the NR2F2 gene variant as de novo. De novo mutations are spontaneous changes in the genetic material present in the fetus, but are not found in the parents. According to the OMIM, this syndrome exhibits clinical variability. Based on these results, we estimated the risk of a couple having another child with this probable pathogenic variant in the NR2F2 gene and CHD to be low. Therefore, there is no contraindication for the couple to pursue another natural pregnancy. International literature also describes the molecular and physiological link between the developing fetal heart and placenta, and the commonality of pathophysiological disorders [21]. Leon R.L. et al. (2022) [22] demonstrated that vascular disorders and chronic forms of inflammation are significantly more common in the placentas of patients with fetal HDs [23].
Histological examination of the placenta revealed a small placenta (310 g, <10th percentile), varicose veins of the placenta and umbilical cord, false knots of the umbilical cord, vessel branching, and a greenish fetal portion. The fetus also exhibited signs of hypoxia (hemorrhage under the visceral pleura and under the epicardium). Based on the results of the pathological examination, we concluded that this pregnant woman with a fetal HD exhibited signs of placental dysfunction. Petit F.G. et al. (2007) [24] showed that changes in the NR2F2 gene lead to disturbances in placental formation and vascularization, resulting in miscarriages on days 10th–12th of pregnancy in mice. This clinical case of a fetal HD warrants special attention. It can be assumed that the causes of antenatal fetal death were disorders in the placenta-fetus system caused by systemic disturbances in the formation of vascular endothelial cells and the fetal heart, associated with a pathogenic variant of the NR2F2 gene.
This observation highlights the importance of timely expanded genetic examination of fetuses with HDs, careful dynamic monitoring of pregnant women, and prompt hospitalization when early signs of placental disorders are detected.
Conclusion
For the first time in the national literature, a clinical observation of a fetal HD associated with a probable pathogenic variant in the NR2F2 gene was documented in a pregnant woman with placental disorders and antenatal fetal death.
The management of patients with fetal HDs necessitates careful and timely genetic examination and counseling of the family, as well as ongoing monitoring of pregnant women. Our clinical case demonstrated that exome sequencing results for the family obtained during the antenatal stage, along with data from global literature and genetic databases, can enhance the monitoring of pregnant women during outpatient care.
References
- Zhang L., Yang Z., Yin Y., Huang W., Yi T., Ping J. et al. Using big data to analyze the vaccination status of children with congenital heart disease in Yinzhou District, China. Hum. Vaccin. Immunother. 2024; 20(1): 2319967. https://dx.doi.org/10.1080/21645515.2024.2319967.
- Скворцов В.В., Тумаренко А.В., Байманкулов С.С. Врожденные пороки сердца. Медицинская сестра. 2017; 7: 14-7. [Skvortsov V.V., Tumarenko A.V., Baimankulov S.S. Congenital heart diseases. Meditsinskaya sestra. 2017; (7):14-7. (in Russian)].
- Linhuan H., Danlei C., Zhiming H., Shu K., Jiayi C., Jiayi P. et al. The use of high-resolution SNP arrays to detect congenital cardiac defects. J. Matern. Fetal Neonatal Med. 2024; 37(1): 2301831. https://dx.doi.org/10.1080/14767058.2024.2301831.
- Федеральная служба государственной статистики (Росстат). Здравоохранение в России. Статистический сборник. М.; 2023. 179c. [Federal State Statistics Service (Rosstat). Healthcare in Russia. Statistical compilation. Moscow; 2023. 179p. (in Russian)].
- Богачевская С.А., Капитоненко Н.А., Богачевский А.Н. Эпидемиологическая характеристика врожденных пороков сердца в России и Дальневосточном федеральном округе за последние 10 лет. Дальневосточный медицинский журнал. 2016; 1: 96-101. [Bogatchevskaia S.A., Kapitonenko N.A., Bogatchevskiy A.N. The epidemiological features of congenital heart diseases in the Russian Federation and the Far Eastern Federal district for the last 10 years. Far Eastern Medical Journal. 2016; (1): 96-101. (in Russian)].
- Щеголев А.И., Туманова У.Н., Шувалова М.П., Фролова О.Г. Сравнительный анализ мертворождаемости в Российской Федерации в 2010 и 2012 г. Российский вестник перинатологии и педиатрии. 2015; 60(3): 58-62. [Shchegolev A.I., Tumanova U.N., Shuvalova M.P., Frolova O.G. Comparative analysis of stillbirth rates in the Russian Federation in 2010 and 2012. Russian Bulletin of Perinatology and Pediatrics. 2015; 60(3): 58-62. (in Russian)].
- van Velzen C.L., Türkeri F., Pajkrt E., Clur S.A., Rijlaarsdam M.E.B., Bax C.J. et al. Pregnancy complications in singleton pregnancies with isolated fetal heart defects. Acta Obstet. Gynecol. Scand. 2016; 95(11): 1273-80. https://dx.doi.org/10.1111/aogs.12955.
- Divanovic A., Bowers K., Michelfelder E., Jaekle R., Newman T., Marcotte M. et al. Intrauterine fetal demise after prenatal diagnosis of congenital heart disease: assessment of risk. Prenat. Diagn. 2016; 36(2): 142-7. https://dx.doi.org/10.1002/pd.4755.
- Пак В.С., Тетруашвили Н.К., Бокерия Е.Л., Шубина Е., Зарецкая Н.В., Большакова А.С., Люшнина Д.Г., Кузнецова М.В., Михайловская Г.В., Саделов И.О., Трофимов Д.Ю. Роль хромосомных аномалий при врожденных пороках сердца плода. Акушерство и гинекология. 2023; 10: 86-93. [Pak V.S., Tetruashvili N.K., Bokeriya E.L., Shubina Je., Zaretskaya N.V., Bolshakova A.S., Lyushnina D.G., Kuznetsova M.V., Mikhailovskaya G.V., Sadelov I.O., Trofimov D.Yu. The role of chromosomal abnormalities in fetal congenital heart defects. Obstetrics and Gynecology. 2023; (10): 86-93. (in Russian)]. https://dx.doi.org/10.18565/aig.2023.220.
- Al Turki S., Manickaraj A.K., Mercer C.L., Gerety S.S., Hitz M.-P., Lindsay S. et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am. J. Hum. Genet. 2014; 94(4): 574-85. https://dx.doi.org/10.1016/j.ajhg.2014.03.007.
- van der Bom T., Zomer A.C., Zwinderman A.H., Meijboom F.J., Bouma B.J.,Mulder B.J.M. The changing epidemiology of congenital heart disease. Nat. Rev. Cardiol. 2011; 8(1): 50-60. https://dx.doi.org/10.1038/nrcardio.2010.166.
- Sun R., Liu M., Lu L., Zheng Y., Zhang P. Congenital heart disease: causes, diagnosis, symptoms, and treatments. Cell. Biochem. Biophys. 2015; 72(3): 857-60. https://dx.doi.org/10.1007/s12013-015-0551-6.
- Geddes G.C., Przybylowski L.F., Ware S.M. Variants of significance: medical genetics and surgical outcomes in congenital heart disease. Curr. Opin. Pediatr. 2020; 32(6): 730-8. https://dx.doi.org/10.1097/MOP.0000000000000949.
- De Backer J., Bondue A., Budts W., Evangelista A., Gallego P., Jondeau G. et al. Genetic counselling and testing in adults with congenital heart disease: A consensus document of the ESC Working Group of Grown-Up Congenital Heart Disease, the ESC Working Group on Aorta and Peripheral Vascular Disease and the European Society of Human Genetics. Eur. J. Prev. Cardiol. 2020; 27(13): 1423-35. https://dx.doi.opg/10.1177/2047487319854552.
- Shikany A.R., Landis B.J., Parrott A., Miller E.M., Coyan A., Walters L. et al. A comprehensive clinical genetics approach to critical congenital heart disease in infancy. J. Pediatr. 2020; 227: 231-8.e14. https://dx.doi.org/10.1016/j.jpeds.2020.07.065.
- Planchais J., Boutant M., Fauveau V., Qing L.D., Sabra-Makke L., Bossard P. et al. The role of chicken ovalbumin upstream promoter transcription factor II in the regulation of hepatic fatty acid oxidation and gluconeogenesis in newborn mice. Am. J. Physiol. Endocrinol. Metab. 2015; 308(10): E868-78. https://dx.doi.org/10.1152/ajpendo.00433.2014.
- Dougherty E.J., Chen L.-Y., Awad K.S., Ferreyra G.A., Demirkale C.Y., Keshavarz A. et al. Inflammation and DKK1-induced AKT activation contribute to endothelial dysfunction following NR2F2 loss. Am. J. Physiol. Lung Cell. Mol. Physiol. 2023; 324(6): L783-98. https://dx.doi.org/10.1152/ajplung.00171.2022.
- Xiaodi L., Ming Y., Hongfei X., Yanjie Z., Ruoyi G., Ma X. et al. DNA methylation at CpG island shore and RXRα regulate NR2F2 in heart tissues of tetralogy of Fallot patients. Biochem. Biophys. Res. Commun. 2020; 529(4): 1209-15. https://dx.doi.org/10.1016/j.bbrc.2020.06.110.
- Ganapathi M., Matsuoka L.S., March M., Li D., Brokamp E., Benito-Sanz S. et al. Heterozygous rare variants in NR2F2 cause a recognizable multiple congenital anomaly syndrome with developmental delays. Eur. J. Hum. Genet. 2023; 31(10): 1117-24. https://dx.doi.org/10.1038/s41431-023-01434-5.
- Bashamboo A., Eozenou C., Jorgensen A., Bignon-Topalovic J., Siffroi J.P., Hyon C. et al. Loss of function of the nuclear receptor NR2F2, encoding COUP-TF2, causes testis development and cardiac defects in 46, XX children. Am. J. Hum. Genet. 2018; 102(3): 487-93. https://dx.doi.org/10.1016/j.ajhg.2018.01.021.
- Mahadevan A., Tipler A., Jones H. Shared developmental pathways of the placenta and fetal heart. Placenta. 2023; 141: 35-42. https://dx.doi.org/10.1016/j.placenta.2022.12.006.
- Leon R.L., Sharma K., Mir I.N., Herrera C.L., Brown S.L., Spong C.Y. et al. Placental vascular malperfusion lesions in fetal congenital heart disease. Am. J. Obstet. Gynecol. 2022; 227(4): 620.e1-620.e8. https://dx.doi.org/10.1016/j.ajog.2022.05.038.
- O’Hare C.B., Mangin-Heimos K.S., Gu H., Edmunds M., Bebbington M., Lee C.K. et al. Placental delayed villous maturation is associated with fetal congenital heart disease. Am. J. Obstet. Gynecol. 2023; 228(2): 231.e1-231.e11. https://dx.doi.org/10.1016/j.ajog.2022.08.013.
- Petit F.G., Jamin S.P., Kurihara I., Behringer R.R., DeMayo F.J., Tsai M.J. et al. Deletion of the orphan nuclear receptor COUP-TFII in uterus leads to placental deficiency. Proc. Natl. Acad. Sci. U. S. A. 2007; 104(15): 6293-8. https://dx.doi.org/10.1073/pnas.0702039104.
Received 01.08.2024
Accepted 10.10.2024
About the Authors
Viktoriia S. Pak, PhD student, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4 Acad. Oparin str., Moscow, Russia, 117997, +7(913)897-28-49,v_pak@oparina4.ru, https://orcid.org/0009-0002-1444-9071
Nana K. Tetruashvili, PhD, Head of the Obstetric Department of Pregnancy Pathology No. 2, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia,
4 Acad. Oparin str., Moscow, Russia, +7(495)438-14-77, n_tetruashvili@oparina4.ru, https://orcid.org/0000-0002-9201-2281
Ekaterina L. Bokerija, PhD, Researcher at the Department of Pathology for Newborn and Prematurely-Born Children No. 2, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4 Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-27-05, e_bokeriya@oparina4.ru, https://orcid.org/0000-0002-8898-9612
Jekaterina Shubina, PhD in Biology, Head of the Laboratory of Genomic Data Analysis, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4 Acad. Oparin str., Moscow, Russia, 117997, +7(495)531-44-44, e_shubina@oparina4.ru, https://orcid.org/0000-0003-4383-7428
Nadezhda V. Zaretskaya, PhD, Head of the Laboratory of Clinical Genetics of the Institute of Reproductive Genetics, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4 Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-24-11, n_zaretskaya@oparina4.ru, https://orcid.org/0000-0001-6754-3833
Anna S. Bolshakova, MD, Geneticist, Department of Clinical Genetics of the Institute of Reproductive Genetics, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia,
4 Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-24-11, a_bolshakova@oparina4.ru, https://orcid.org/0000-0002-7508-0899
Daria G. Lyushnina, PhD student, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4 Acad. Oparin str., Moscow, Russia, 117997, +7(906)308-60-78,
d_lyushnina@oparina4.ru, https://orcid.org/0009-0004-3160-8737
Dmitry N. Maslennikov, MD, Geneticist at the Laboratory of Genomic Data Analysis of the Institute of Reproductive Genetics, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4 Acad. Oparin str., Moscow, Russia, 117997, d_maslennikov@oparina4.ru, https://orcid.org/0000-0001-5916-0672
Taisiya O. Kochetkova, Biologist at the Laboratory of Molecular and Genetic Methods of the Institute of Reproductive Genetics, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4 Acad. Oparin str., Moscow, Russia, 117997, t_kochetkova@oparina4.ru, https://orcid.org/0000-0003-0215-3636
Dmitry Yu. Trofimov, Dr. Bio. Sci, Professor of the RAS, Corresponding Member of the RAS, Director of the Institute of Reproductive Genetics, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4 Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-49-51, d_trofimov @oparina4.ru, https://orcid.org/0000-0002-1569-8486
Similar Articles

