
Понятие о тромботической микроангиопатии и тромботической тромбоцитопенической пурпуре
В настоящее время тромботическая тромбоцитопеническая пурпура (ТТП) и гемолитико-уремический синдром (ГУС) рассматриваются как проявления тромботической микроангиопатии. Тромботическая микроангиопатия морфологически проявляется утолщением стенок сосудов микроциркуляторного русла (преимущественно капилляров и артериол), отеком и слущиванием эндотелиальных клеток от базальной мембраны, образованием тромбоцитарных сгустков и частичной или полной обструкцией просвета пораженного сосуда, при этом периваскулярное воспаление не характерно, а тромбы состоят почти исключительно из тромбоцитов. Обструкция просвета сосудов приводит к развитию ишемии и инфарктов органов. Характерным признаком тромботической микроангиопатии является тромбоцитопения и гемолитическая анемия, что связано с потреблением и разрушением тромбоцитов и эритроцитов в микроциркуляторном русле [1, 2].
ТТП характеризуется тромбоцитопенией, микроангиопатической анемией, лихорадкой, поражением почек и неврологической симптоматикой. Неврологические проявления ТТП крайне разнообразны и варьируют от небольших нарушений поведения и затуманенности сознания до выраженных сенсорно-моторных нарушений, афазии, судорог и комы. Кроме того, при ТТП могут наблюдаться боли в животе, панкреатит, гематурия, нарушения сердечного ритма, нарушения зрения. Для ГУС также характерна тромбоцитопения и микроангиопатическая гемолитическая анемия, но с преимущественным поражением почек.
 После внедрения терапии свежезамороженной плазмой и значительного снижения летальности у больных ТТП удалось проследить дальнейшую судьбу этих пациентов, и стало очевидно, что этиология ТТП характеризуется значительным разнообразием, как и дальнейшее течение этого заболевания. В настоящее время выделяют наследственную (семейную, врожденную) форму ТТП, которая носит название синдрома Апшоу–Шульмана и обусловлена генетическим дефектом протеазы vWF – ADAMTS-13, и приобретенную форму ТТП, обусловленную формированием антител к ADAMTS-13 или ее ингибитора [3]. Мутации в гене ADAMTS-13 вызывают значительное снижение уровня этого фермента в плазме крови или выраженное нарушение его активности. При тяжелом генетически обусловленном дефиците ADAMTS-13 эпизоды ТТП могут начаться с раннего детства, однако у ряда больных заболевание может долго себя не проявлять вплоть до воздействия какого-либо сильного провоцирующего фактора. Например, триггером для развития первого эпизода ТТП у таких больных может стать беременность, различные инфекционные заболевания и септические состояния, сопровождающиеся массивным выбросом провоспалительных цитокинов, а также прием оральных контрацептивов, так как содержащиеся в них эстрогены стимулируют выброс ультравысокомолекулярных мультимеров vWF из эндотелиоцитов. У ряда больных с выраженным наследственным дефицитом ADAMTS-13 (активность ADAMTS-13 в плазме крови менее 5–10%) ТТП принимает хроническое рецидивирующее течение с рождения, а ведущим синдромом становится прогрессирующая почечная недостаточность. При редкой врожденной форме ТТП рецидивы могут возникать каждые 3–4 недели. Такую форму заболевания часто называют хронической рецидивирующей ТТП. У двух третей больных с относительно более распространенной приобретенной формой ТТП в случае успешной терапии повторные эпизоды не возникают, тогда как у трети больных развиваются рецидивы [4]. Когда именно разовьется рецидив, предсказать невозможно. Период ремиссии может длиться от нескольких дней до десятков лет, однако наиболее часто рецидив развивается в течение года после первого эпизода ТТП. Триггером к развитию рецидива могут служить беременность, хирургическое вмешательство, инфекция, вакцинация.
После внедрения терапии свежезамороженной плазмой и значительного снижения летальности у больных ТТП удалось проследить дальнейшую судьбу этих пациентов, и стало очевидно, что этиология ТТП характеризуется значительным разнообразием, как и дальнейшее течение этого заболевания. В настоящее время выделяют наследственную (семейную, врожденную) форму ТТП, которая носит название синдрома Апшоу–Шульмана и обусловлена генетическим дефектом протеазы vWF – ADAMTS-13, и приобретенную форму ТТП, обусловленную формированием антител к ADAMTS-13 или ее ингибитора [3]. Мутации в гене ADAMTS-13 вызывают значительное снижение уровня этого фермента в плазме крови или выраженное нарушение его активности. При тяжелом генетически обусловленном дефиците ADAMTS-13 эпизоды ТТП могут начаться с раннего детства, однако у ряда больных заболевание может долго себя не проявлять вплоть до воздействия какого-либо сильного провоцирующего фактора. Например, триггером для развития первого эпизода ТТП у таких больных может стать беременность, различные инфекционные заболевания и септические состояния, сопровождающиеся массивным выбросом провоспалительных цитокинов, а также прием оральных контрацептивов, так как содержащиеся в них эстрогены стимулируют выброс ультравысокомолекулярных мультимеров vWF из эндотелиоцитов. У ряда больных с выраженным наследственным дефицитом ADAMTS-13 (активность ADAMTS-13 в плазме крови менее 5–10%) ТТП принимает хроническое рецидивирующее течение с рождения, а ведущим синдромом становится прогрессирующая почечная недостаточность. При редкой врожденной форме ТТП рецидивы могут возникать каждые 3–4 недели. Такую форму заболевания часто называют хронической рецидивирующей ТТП. У двух третей больных с относительно более распространенной приобретенной формой ТТП в случае успешной терапии повторные эпизоды не возникают, тогда как у трети больных развиваются рецидивы [4]. Когда именно разовьется рецидив, предсказать невозможно. Период ремиссии может длиться от нескольких дней до десятков лет, однако наиболее часто рецидив развивается в течение года после первого эпизода ТТП. Триггером к развитию рецидива могут служить беременность, хирургическое вмешательство, инфекция, вакцинация.
В настоящее время принята следующая классификация тромботических микроангиопатий (табл. 1).
Молекулярные основы патогенеза тромботической микроангиопатии
Moake и соавт. в 1982 г. впервые выявили аномальные мультимерные комплексы vWF у пациентов с ТТП и сделали предположение о возможной роли vWF в патогенезе ТТП [5]. Характерным признаком ТТП является дефицит плазменной протеазы, расщепляющей мультимеры vWF – ADAMTS-13. При семейных формах ТТП наблюдается наследственный дефект этого фермента, в то время как приобретенные формы ТТП характеризуются наличием антител-ингибиторов vWF-протеазы [6].
Фактор Виллебранда представляет собой высокомолекулярный мультимер, образующийся при полимеризации мономерных субъединиц с молекулярной массой 225 кДа в эндотелиальных клетках и мегакариоцитах и накапливающийся в тельцах Weibel-Palade в эндотелиальных клетках и а-гранулах тромбоцитов. Эти ультравысокомолекулярные мультимеры vWF (ULVWF) секретируются активированными эндотелиоцитами наподобие «лент». В норме эти «ленты» высокомолекулярных комплексов vWF сразу после экспрессии на плазматической мембране подвергаются распаду на фрагменты с Мг 189, 176 и 140 кДа под действием плазматической металлопротеазы ADAMTS-13 и, следовательно, в циркулирующей крови не обнаруживаются [3]. Физиологическая роль мультимера vWF заключается в обеспечении адгезии тромбоцитов к субэндотелиальному матриксу в условиях повреждения сосуда и гемодинамического стресса. Низкомолекулярные фрагменты vWF, циркулирующие в системном кровотоке, обладают слабой способностью к связыванию с тромбоцитами и не проявляют гемостатическую активность. В то время как аффинность отдельных субъединиц vWF к тромбоцитам чрезвычайно мала, мультимеры vWF обеспечивают одновременно множество участков связывания с рецепторами Ib-тромбоцитов, что позволяет значительно увеличить силу взаимодействия vWF-тромбоцитов. Так, аффинность высокомолекулярной формы vWF к тромбоцитам в 10 раз превышает таковую у отдельных субъединиц vWF [7]. Мультимерные «ленты» ULVWF могут фиксироваться на поверхности мембран эндотелиальных клеток при помощи Р-селектина, который секретируется из телец Weibel-Palade одновременно с ULVWF. В результате в условиях относительного или абсолютного дефицита ADAMTS-13 микрососуды перекрываются гигантскими ультравысокомолекулярными vWF, на которых оседает возрастающее количество тромбоцитов, образующих блокирующие микрососудистое русло тромбоцитарные тромбы. Одним из факторов, которые стимулируют выброс ULVWF из эндотелиальных клеток, являются провоспалительные цитокины. В связи с этим состояния, сопровождающиеся активацией процессов системного воспаления, в том числе такие осложнения беременности, как преэклампсия, могут стать стимулом к развитию тромботической микроангиопатии.
В норме у здоровых людей активность ADAMTS-13 варьирует в значительных пределах от 50 до 170%. Cнижение активности vWF-протеазы ниже нормы (менее 50%) наблюдается в течение третьего триместра беременности, при циррозе печени, диссеминированных опухолях и воспалительных заболеваниях. У пациентов, переживших ТТП, мультимерные комплексы vWF обнаруживаются лишь в острую фазу заболевания и не обнаруживаются в кровотоке после выздоровления. Возможно, при массивном повреждении эндотелия происходит значительный выброс vWF из гранул; при этом возникает относительная недостаточность металлопротеазы. Однако у пациентов, страдающих рецидивирующей формой ТТП, мультимеры vWF в кровотоке выявляются постоянно, как в острую фазу заболевания, так и в период ремиссии. Такая рецидивирующая форма заболевания чаще является наследственной и обусловлена отсутствием или дефицитом протеазы ADAMTS-13. Так, у большинства пациентов с семейной формой ТТП активность ADAMTS-13 в плазме крови составляет 5–10%, в то время как у большинства пациентов с приобретенной идиопатической ТТП подобное снижение активности ADAMTS-13 выявляется только в период рецидивов [8].
В отличие от сериновых протеаз, у металлопротеазы vWF в норме не обнаруживается плазменный ингибитор. Если для большинства металлопротеаз период полужизни измеряется секундами и минутами, для vWF-протеазы этот показатель составляет 2–4 дня [9]. Поэтому у пациентов с рецидивирующей ТТП и наследственным дефектом vWF-протеазы при применении плазмы, содержащей vWF-протеазу, может быть достигнута ремиссия заболевания. При дефиците vWF, обусловленной наличием ингибитора, целью плазмафереза является удаление патогенных IgG; возможно также применение иммунносупрессивных препаратов (глюкортикоидов, винкристина). Ингибитор vWF-протеазы вновь появляется в крови через 3 месяца после лечения.
Антитела IgG к ADAMTS-13 выявляются у 44–94% пациентов с приобретенной формой ТТП [9].
Их уровень обычно возрастает в острый период или во время рецидива ТТП, тогда как в период ремиссии антитела к ADAMTS-13 в плазме крови у таких больных могут не выявляться. Постоянное обнаружение антител более характерно для больных с частыми рецидивами заболевания, для которых также характерно выявление выраженного дефицита ADAMTS-13. Причинами такого транзиторного выявления антител к ADAMTS-13 может быть, с одной стороны, недостаточная чувствительность доступных в настоящее время диагностических методик, а с другой стороны, дефекты иммунной регуляции, на фоне которых возможно возникновение интермиттирующего дефицита ADAMTS-13 под действием различных провоцирующих факторов (беременность, инфекционные заболевания).
Лабораторная диагностика тромботической микроангиопатии
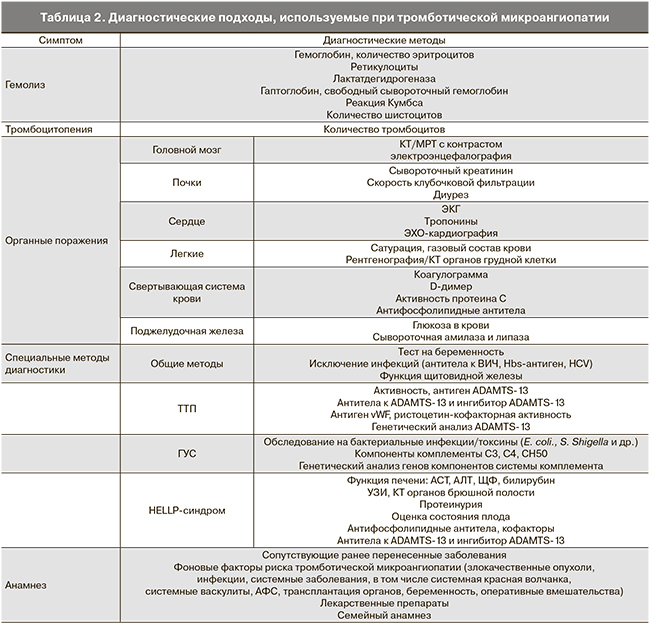
Типичными лабораторными проявлениями ГУС/ТТП являются тромбоцитопения и гемолитическая анемия [10]. Характерно увеличение содержания лактатдегидрогеназы в сыворотке, что обусловлено активацией гемолиза, а также является признаком тканевой ишемии. Другими признаками гемолиза и перераздражения эритроцитарного ростка служат повышение билирубина (преимущественно непрямого), количества свободного гемоглобина и ретикулоцитов в периферической крови. Характерными признаками микроангиопатической природы гемолиза является обнаружение фрагментов эритроцитов – шистоцитов и отрицательная реакция Кумбса. Причиной образования шистоцитов является резкое сужение сосудов, создающее условия для гемодинамического стресса, обусловливающего фрагментацию эритроцитов. Диагностические подходы, используемые при тромботической микроангиопатии, описаны в табл. 2.
Особенности тромботической микроангиопатии, ассоциированной с беременностью
Беременность по праву считается одним из важнейших триггерных факторов для развития ТТП. В 12–31% случаев ГУС/ТТП развивается во время беременности или в раннем послеродовом периоде [1, 10]. Заболеваемость ТТП во время беременности составляет 1 на 25–100 000 [1]. До внедрения в клиническую практику плазмафереза материнская смертность при ГУС/ТТП составляла 95%, а перинатальная – 80% [10]. Во время беременности наблюдается, с одной стороны, прогрессивное повышение уровня vWF, вероятно, под действием эстрогенов, а с другой, снижение активности ADAMTS-13, что может быть обусловлено повышенным потреблением этого фермента, действие которого направлено на разрушение избыточных количеств ультравысокомолекулярных мультимеров vWF, экспрессируемых активированным эндотелиоцитами. Таким образом, беременность может стать провоцирующим фактором для развития ТТП при генетическом дефекте ADAMTS-13. Кроме того, ТТП во время беременности встречалась и у пациенток с антителами к ADAMTS-13. Был описан интереснейший клинический случай: у 23-летней женщины в течение 73 месяцев было четыре беременности, заканчивавшиеся самопроизвольными абортами в первом триместре, после чего у нее развивались эпизоды ТТП, регрессировавшие на фоне лечения кортикостероидами и плазмаферезом. После имплантации противозачаточного средства новых эпизодов ТТП не отмечалось [11]. В настоящее время критериями для постановки диагноза из 5 характерных признаков этого заболевания служат только тромбоцитопения и микроангиопатическая гемолитическая анемия. Клинически ТТП/ГУС в этом случае часто бывает трудно отличить от преэклампсии, эклампсии и HELLP-синдрома, для которых также характерно развитие тромоцитопении и микроангиопатической гемолитической анемии. Ситуация осложняется еще и тем, что HELLP-синдром и экламптические судороги могут развиваться и на фоне нормального артериального давления [12].
Таким образом, развитие тромботической микроангиопатии характерно для HELLP-синдрома, ТТП, ГУС, а также является одним из проявлений катастрофического АФС. Это свидетельствует о едином механизме патогенеза этих заболеваний. Рядом исследователей описаны случаи возникновения HELLP-синдрома у женщин с АФС, что лишний раз подтверждает роль патологии гемостаза как предрасполагающего фактора к возникновению HELLP-синдрома [13]. Следует также учитывать, что HELLP-синдром может быть первым проявлением АФС. Появились данные о роли антител к ADAMTS-13 в качестве причины тромбоцитопении у больных системной красной волчанкой, что может быть одним из критериев неблагоприятного прогноза заболевания у таких пациентов [14]. O. Pourrat и соавт. (2013) описывают взаимосвязь между дефицитом ADAMTS-13 и развитием HELLP-синдрома [15]. Более того, появились интересные данные о том, что антитела к ADAMTS-13 могут формироваться в условиях АФС, что может являться важнейшим фактором развития тромботических и акушерских осложнений [16]. Таким образом, антитела к ADAMTS-13 и дисфункция ADAMTS-13 могут развиваться и при других аутоиммунных заболеваниях, помимо приобретенной ТПП, в частности, в условиях АФС. В связи с этим мы считает, что у женщин с HELLP-синдромом необходим анализ на антифосфолипидные антитела и оценка активности ADAMTS-13 и наличия его ингибиторов.
Принципы терапии ГУС/ТТП
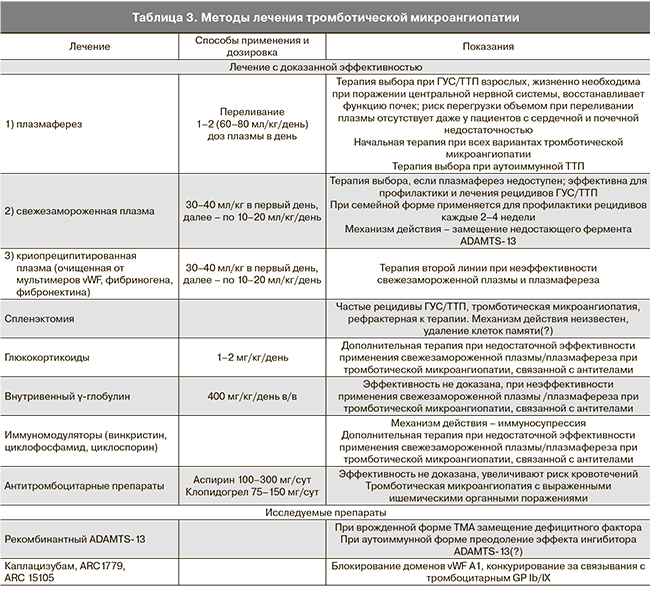
Терапией выбора при ТТП/ГУС является применение свежезамороженной плазмы или плазмафереза. Применение обменного переливания плазмы позволяет снизить уровень смертности при ГУС/ТТП с 80 до 10%. Целью применения плазмафереза является возмещение уровня vWF-протеазы, удаление антител, блокирующих активность ADAMTS-13, провоспалительных цитокинов, компонентов комплемента из системного кровотока, а также возмещение дефицита естественных антикоагулянтов, что особенно важно при сочетании ТТП/ГУС с генетическими тромбофилиями и АФС. При наследственной форме, обусловленной гомозиготной или двумя гетерозиготными мутациями ADAMTS-13, эффективно применение свежезамороженной плазмы, а для профилактики рецидивов заболевания переливание плазмы необходимо применять раз в 2–3 недели [17]. У больных с приобретенной формой ТТП, у которых дефицит ADAMTS-13 в большинстве обусловлен не абсолютным отсутствием этого белка, а блокадой его активности вследствие циркуляции аутоантител, только переливания свежезамороженной плазмы может оказаться недостаточно, так как имеющиеся антитела будут блокировать и вновь поступающее в организм количество ADAMTS-13. Тем не менее, инфузия свежезамороженной плазмы у больных с приобретенной формой ТТП должна быть начата сразу после появления подозрения на ТТП при отсутствии возможности начать плазмаферез в экстренном порядке или до того, как будет уточнен диагноз [18].
Помимо терапии плазмой, применяется целый ряд методов и лекарственных препаратов, однако эффективность большинства их них не доказана (табл. 3). Эти методы направлены на подавление синтеза аутоантител и применяются у больных с приобретенной ТТП и аутоантителами к ADAMTS-13 при отсутствии эффекта от стандартной терапии с применением плазмафереза и свежезамороженной плазмы. Возможные варианты терапии включают высокие дозы глюкокортикоидов, ритуксимаб (моноклональное антитела к CD20 на В-лимоцитах), в комбинации с циклофосфамидом, циклоспорином, спленэктомией. Так, появились данные о повышении эффективности терапии приобретенной ТТП при одновременном применении плазмафереза и глюкокортикоидов [19]. Рекомендуют начинать преднизолон внутривенно в дозе 200 мг в день сразу же после установления диагноза ТТП и продолжать лечение с постепенным снижением дозы вплоть до выздоровления [20].
Применение глюкокортикоидов до и после родов способствует уменьшению тяжести HELLP-синдрома, потребности в гемотрансфузии и позволяет продлить беременность на 24–48 ч, что важно для профилактики респираторного дистресс-синдрома новорожденных [21]. Предполагается, что применение глюкокортикоидов может способствовать восстановлению функций эндотелия, блокаде аутоиммунных патогенетических механизмов, предотвращать внутрисосудистое разрушение эритроцитов и тромбоцитов и прогрессирование системного воспалительного ответа. В частности, рассматривая HELLP-синдром как вариант приобретенной тромботической микроангиопатии, эффективность глюкокортикоидов можно объяснить блокирующим эффектом в отношении антител к ADAMTS-13 и антифосфолипидных антител. Однако вслед за улучшением клинической картины, отмечаемым в течение 24–48 ч на фоне применения глюкокортикоидов, может возникнуть так называемый «ребаунд»-феномен, проявляющийся ухудшением состояния беременной. Таким образом, введение глюкокортикоидов не предотвращает полностью развитие патологического процесса, а лишь кратковременно улучшает клиническую картину, создавая условия для более успешного родоразрешения.
Важно отметить, что всем пациентам с ТТП, несмотря на выраженную тромбоцитопению, необходимо проведение тромбопрофилактики низкомолекулярным гепарином. Несмотря на выраженную тробоцитопению, переливание тромбоцитов показано только у пациентов с угрожающими жизни кровотечениями, так как это может спровоцировать прогрессирование тромботической микроангиопатии.
Заключение
Открытия последних лет, связанные с изучением молекулярных механизмов тромботический микроангиопатии, позволяют сделать вывод, что эта патология гораздо более распространена, чем было принято считать ранее. Важнейшим триггером к развитию как ТТП, так и других вариантов тромботической микроангиопатии является беременность. Причинами тому могут быть физиологическое повышение уровня фактора фон Виллебранда, характерное для беременности, активное потребление и истощение запасов ADAMTS-13, что может усугублять ранее скрытый, умеренный генетически обусловленный дефицит этого фермента. Кроме того, последние данные указывают на взаимосвязь между циркуляцией антифосфолипидных антител и приобретенным дефицитом ADAMTS-13. В частности, такие механизмы могут играть важную роль в патогенезе тяжелых плацентарных осложнений беременности, в том числе преэклампсии, HELLP-синдрома. В настоящее время критерии для установления диагноза тромботической микроангиопатии значительно расширены. Эту патологию следует исключать у всех пациентов, у которых выявляется тромбоцитопения в сочетании с гемолитической анемией. Своевременная диагностика тромботической микроангиопатии имеет колоссальное значение для выбора тактики лечения, в том числе может полностью изменить подход к терапии пациенток с преэклампсией и HELLP-синдромом. Возможно, более глубокое изучение патогенеза тромботической микроангиопатии позволит разработать более чувствительные и специфичные методы диагностики этого патологического процесса и разработать эффективные методы его терапии.


