
Согласно определению Европейского общества репродукции человека (ESHRE), преждевременная недостаточность яичников (ПНЯ) – клинический синдром, ассоциированный с вторичной аменореей в сочетании с высоким уровнем ФСГ (более 25 мМЕ) у женщин в возрасте до 40 лет [1].
В исследовании Study of Women's Health Across the Nation (SWAN) от 2003 г., частота ПНЯ среди представительниц европеоидной расы составляет 1%, афроамериканок – 1,4%, латиноамериканок – 1,4%, китаянок – 0,5% и японок – 0,1% [2]. По данным масштабного метаанализа 2019 г., распространенность ПНЯ прогрессивно возрастает и к настоящему времени достигла 3,7% среди европейской популяции женщин в возрасте до 40 лет [3]. Еще более выраженный рост ПНЯ отмечен в китайской популяции, на что указывает широкомасштабное исследование 2014 г., включающее 36 402 женщин в возрасте от 49 до 66 лет, в котором частота ПНЯ достигла 2,76% (т.е. увеличилась за последние 12 лет в 6 раз) [4].
Патогенетической основой развития ПНЯ является нефизиологическое по отношению к возрасту женщины снижение тотального овариального резерва, нарушение процессов рекрутирования фолликулов или их ускоренный апоптоз, а также развитие феномена преждевременной лютеинизации фолликулов [5, 6].
Преждевременному истощению овариального резерва могут способствовать хромосомные и генетические аномалии, эпигенетические причины, аутоиммунная патология, развивающаяся в рамках аутоиммунного полигландулярного синдрома преимущественно II типа, а также инфекционно-токсические заболевания [7].
В более ранних исследованиях генез ПНЯ основывался на выявлении только хромосомных аномалий на основе цитогенетического анализа. При использовании стандартного цитогенетического исследования метода Seabright (G-окраски) при ПНЯ наиболее часто встречались числовые нарушения в кариотипе, по типу Х моно-, ди- и трисомии, а также различные варианты хромосомного мозаицизма и структурных нарушений (делеции, транслокации, инверсии). На долю вышеуказанных нарушений приходится от 10 до 15% в этиологической структуре ПНЯ, однако, по данным Жахур и соавт., этот показатель не превышает 7,7% [1, 8].
В настоящее время наравне с цитогенетическими методами используется технология сравнительной геномной гибридизации (Comparative Genomic Hybridization, CGH), которая применяется для анализа вариаций числа копий (CNV), затрагивающих Х-сцепленные гены, которые ассоциированы с несиндромальными формами ПНЯ. При синдромальных формах, как правило, CNV обнаруживают на аутосомах [9]. CGH позволяет выявить как числовые нарушения хромосом во всем геноме, так и их перестройки, а также характеризуется улучшенным разрешением на 5–10 мегабаз по сравнению с более традиционными методами цитогенетического анализа Гимза-бэнды и флуоресцентной гибридизации in situ (FISH), которые ограничены разрешением используемого микроскопа.
Перед клиницистами встает правомерный вопрос: может ли CGH с высоким разрешением образцов ДНК улучшить диагностику ПНЯ на основе выявления новых генов-кандидатов и уточнить распространенность соматической хромосомной нестабильности среди женщин с идиопатической формой заболевания?
В исследовании Norling A et al., включающем 26 пациенток с ПНЯ, впервые при использовании метода CGH в одном случае был выявлен патогенный вариант гена GDF9, а также идентифицировано несколько новых генов-кандидатов – DNAH6, TSPYL6, SMARCC1, CSPG5 и ZFR2 [10]. В 2019 г. на основе использования вышеуказанной методики на когорте из 67 пациенток с ПНЯ было показано, что в 47,8% случаев выявлены как известные, так и вновь идентифицированные (TP63 и VLDLR) потенциально патогенные CNV, способствующие развитию ПНЯ [11].
Частота хромосомной нестабильности с использованием метода CGH при ПНЯ была изучена в исследовании Katari S. et al., при этом в 18% (3/16) случаев выявлены патогенные CNV. Хромосомные перестройки по типу дупликации 16p12.3 обнаружены в одном случае и в двух – делеция Xq28, что свидетельствует о недостаточной репарации ДНК [12].
Таким образом, в клинической цитогенетике, в рамках акушерской практики, метод CGH используется для преимплантационной генетической диагностики. Однако на основе вышеизложенного метод может быть рекомендован гинекологам-эндокринологам для выявления этиологических причин, лежащих в основе преждевременного снижения овариального резерва.
Несмотря на современные диагностические возможности, в большинстве случаев точно выявить этиологию данного заболевания крайне сложно, в связи с чем, частота идиопатической формы в структуре ПНЯ до последнего времени составляла более 50% [5, 6].
На долю генетических причин, объясняющих первоначальное снижение тотального овариального резерва или ускоренный процесс атрезии примордиальных фолликулов, по данным зарубежных авторов, приходится 20–25%. Согласно данным российских авторов, ПНЯ в ряде случаев можно рассматривать как мультифакторную патологию, в генезе которой в 63% случаев выявляются сочетанные молекулярно-генетические и эпигенетические нарушения преимущественно на Х-хромосоме. К этим нарушениям относятся любые отклонения числа CGG повторов в гене FMR1 по отношению к его нормальному диапазону (28–36), «укорочение» аллеля (менее 24 CAG повтора) в гене AR, неслучайная инактивация Х хромосомы [13].
В последние годы выявлено 70 и более генов-кандидатов, на основе которых пытаются объяснить сложный генез формирования ПНЯ. Согласно клиническому наблюдению Шамиловой Н.Н. за 200 пациентками с ПНЯ показано, что для них характерно своевременное менархе, отсутствие фенотипических стигм и регулярный ритм менструаций. До возраста 27–32 лет в 46,5% случаев спонтанно наступали беременности, заканчивающиеся живорождением в 33,6%, в результате чего некоторые врачи игнорируют генетические причины нарушений фолликулогенеза при преждевременном старении яичников [13]. Для разрешения этой дискуссии следует напомнить, что патологические варианты генов, выявляемые у пациенток с ПНЯ, как правило, гетерозиготные, в связи с чем их клинические симптомы могут появляться на втором-третьем десятилетии жизни.
В будущем в результате широкого внедрения секвенирования нового поколения (NGS) в корне изменится подход к понятию «идиопатическая форма ПНЯ» и станет возможной расшифровка ранее неизвестных патогенных вариантов в генах, отвечающих за различные этапы оо- и фолликулогенеза.
В последние годы в литературе представлен ряд крупных молекулярно-генетических исследований, на основании которых выделена панель наиболее значимых генов-кандидатов, принимающих участие в генезе развития ПНЯ: NANOS3, FIGLA, FOXO3, NR5A1, NOBOX, BMP15, GDF9, PGRMCI, PTEN, BRCA1,2 (таблица) [14, 15]. Данные гены отвечают за ключевую роль в раннем фолликулогенезе, в том числе формирование первичных фолликулов и зоны пеллюцида, их рост до ФСГ-зависимой стадии, ингибирование экспрессии м-РНК-рецепторов ФСГ в гранулезных клетках, тем самым предотвращая преждевременную лютеинизацию фолликулов, а также за ранний стероидогенез и регуляцию процессов апоптоза в примордиальных фолликулах [16].
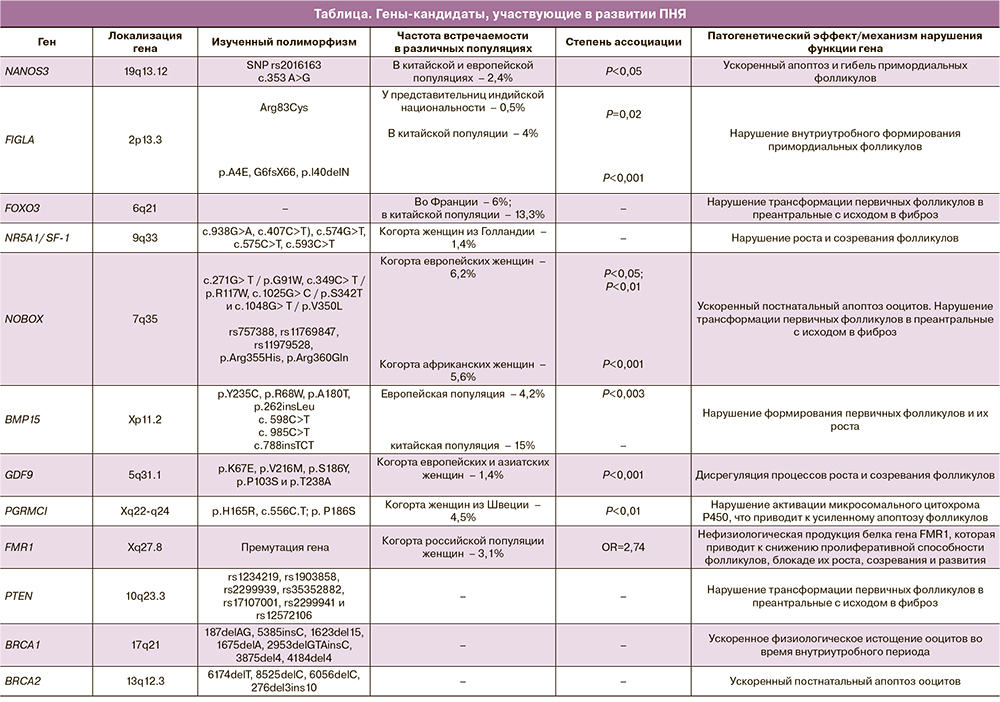
Ген NANOS – семейство генов, участвующее в формировании и развитии примордиальных фолликулов. Существует три гомолога этого гена: NANOS1 (10q26.11), NANOS2 (19q13.32) и NANOS3 (19p13.13) [17]. Ген NANOS3 экспрессируется в яичниках и играет важную роль в образовании и поддержании герминативных стволовых клеток (оогоний), предотвращая их преждевременное вступление в дифференцировку, также отвечая за оогенез, и на этапе миграции зародышевых клеток блокирует процесс их апоптоза. В дальнейшем он участвует в пролиферации примордиальных фолликулов яичника. Поломки в данном гене способствуют бесконтрольному процессу апоптоза и гибели примордиальных фолликулов, приводя к преждевременному истощению фолликулярного пула в яичнике [18]. При ПНЯ у представительниц китайской и европейской популяции патогенный вариант гена NANOS выявлен в 2,4% случаев [1].
Ген FIGLA (специфичный для фолликулогенеза транскрипционный фактор), локализуется на коротком плече Х-хромосомы. Играет важную роль в образовании первичных фолликулов и координирует экспрессию генов зоны пеллюцида. Экспериментально доказано, что наличие мутации в гене FIGLA приводит к блокаде внутриутробного формирования примордиальных фолликулов и в последующем – к постнатальной быстрой потере тотального овариального резерва у мышей. Zhao H. et al. в 4 случаях у 100 китаянок с ПНЯ обнаружили 3 варианта миссенс-мутаций в данном гене (р.А4Е у двух женщин, р.G6fsX66 и p.l40delN). Авторы пришли к заключению, что у пациенток со спорадическими формами ПНЯ одна из ведущих ролей (4%) принадлежит гену FIGLA [19]. В 2015 г. выявлен новый редкий вариант мутации Arg83Cys в изучаемом гене у представительниц индийской национальности (0,5%) [20].
Ген FOXO3 картирован на хромосоме 6 в локусе q21. Транскрипционный фактор FOXO3 отвечает за регуляцию процессов рекрутирования фолликулов. Удаление FOXO3 у мышей приводит к формированию ПНЯ из-за неконтролируемого ускоренного рекрутинга фолликулов [21]. В популяции французских и китайских женщин частота встречаемости патогенных вариантов гена FOXO3 достигает 6 и 13,3% соответственно, приводя к ускоренной потере тотального овариального резерва. FOXO3 оказывает блокирующее влияние на продуцируемые ооцитом гены GDF9, BMP15 и FSHR (ген рецептора к ФСГ), которые в свою очередь блокируют процесс рекрутинга, подавляя нормальную пролиферацию клеток гранулезы [21].
Ген NR5A1 относится к подсемейству ядерных рецепторов 5, группа А, член 1 – данный ген кодирует ядерный рецептор, экспрессия которого может быть выявлена на ранней стадии развития эмбриона в бипотентных гонадах. Ген NR5A1 кодирует ядерный рецептор, регулирующий транскрипцию множества генов, участвующих в стероидогенезе и мужской половой дифференциации. Таким образом, он регулирует продукцию АМГ, гонадотропинов, цитохрома 450, CYP11, а также генов, кодирующих стероидную гидроксилазу и ароматазу, и стероидогенного регуляторного белка (STAR). Инактивация данного гена у экспериментальных мышей приводит к гипоплазии яичников и бесплодию [16]. Полиморфизм NR5A1 является патогенетической основой формирования ПНЯ, дисгенезии гонад и синдрома нечувствительности к андрогенам (тестикулярной феминизации), что подтверждено рядом клинических наблюдений [22]. В семьях у пациенток с ПНЯ идентифицировано 19 различных патогенных вариантов в данном гене [23].
Janse F. et al., обследовав когорту, включающую 356 голландских женщин с ПНЯ, в 1,4% случаев выявили различные патогенные варианты в гене NR5A1 [24]. Таким образом, становится понятным, что мутантный белок данного гена приводит к изменению трансактивационной активности в промоторе гена, что, в свою очередь, нарушает рост и созревание фолликулов.
Ген NOBOX играет ведущую роль в формировании раннего фолликулогенеза и экспрессируется в ооците, кодируя ооцит-специфические факторы транскрипции. Данный ген впервые идентифицирован Suzumori N. et al. в 2002 г. [25]. Ген NOBOX, известен как первый аутосомный ген-кандидат, картированный на 7 хромосоме в локусе q35. Его дефицит у мышей нарушает ранний фолликулогенез, приводя к ускоренному постнатальному апоптозу ооцитов, блокируя трансформацию первичных фолликулов в преантральные, способствуя их фиброзированию с исходом в ПНЯ [26]. В 2007 г. Qin Y. et al. впервые описали патогенный вариант гена NOBOX у женщин с ПНЯ [27]. Ученые из Франции на широкомасштабной когорте европейских и африканских пациенток продемонстрировали частоту встречаемости спорадической формы ПНЯ в пределах 6,2 и 5,6% соответственно, в то время как у 200 представительниц китайской популяции с ПНЯ подобного патогенного варианта данного гена выявлено не было ни в одном случае [28, 29].
Для понимания генеза ПНЯ необходимо рассмотреть роль ооцит-специфического фактора, гена BMP15 (костный морфогенетический белок 15), являющегося членом суперсемейства трансформирующего фактора роста (TGF)-β, который выполняет одну из ключевых ролей на ранних этапах фолликулогенеза. Ген фактора ВМР15 картируется на X-хромосоме в локусе Хp11.2 [30]. Среди его разнообразных функций следует упомянуть экспрессию ооцитом, необходимую для формирования первичных фолликулов и начала их роста. Впоследствии за счет ингибирования экспрессии мРНК рецепторов ФСГ в гранулезных клетках он блокирует позднюю ФСГ-зависимую стадию развития фолликулов и ФСГ-зависимую продукцию прогестерона, тем самым предотвращая преждевременную их лютеинизацию. Впервые роль BMP15 в генезе ПНЯ была доказана на основе выявленной миссенс-мутации р.Y235C у двух сестер с ПНЯ [31]. В дальнейшем различные патологические варианты этого гена были идентифицированы у представительниц европейской, индийской и китайской популяций с частотой от 4,2 до 15% [31–33].
В России был проведен анализ полиморфных вариантов данного гена в позициях: 202C> T (Arg68Trp, rs104894763), 538G> A (А180T, rs3897937) и 905G> A (rs104894767) у 101 пациентки с ПНЯ. Сравнение частоты встречаемости данных нуклеотидных замен у обследуемой когорты и 101 женщины со своевременной менопаузой не выявило достоверной разницы, что указывает на возможно незначимую роль данных полиморфизмов в генезе ПНЯ для россиянок [13].
Ген GDF9 (фактор дифференцировки роста) экспрессируется также ооцитом, гомологичен гену BMP15 и подобно ему является членом семейства генов TGF. Экспериментально доказано, что у нокаутных по гену GDF9 мышей в результате торможения пролиферации гранулезных клеток и потери чувствительности к ФСГ полностью блокируются процессы роста и созревания фолликулов. Таким образом, нормальное функционирование GDF9 имеет решающее значение в процессе селекции доминантного фолликула [34, 35]. Первые полномасштабные исследования по изучению полиморфизма гена GDF9 (2005–2007), проведены на когорте европейских и азиатских женщин с ПНЯ (629 пациенток) – частота патогенных вариантов составила 1,4%. Полиморфизм гена, описанный в европейских и азиатских группах женщин, кроме представительниц Японии и Новой Зеландии, относятся к миссенс-мутациям и встречаются только в гетерозиготном состоянии с частотой до 4%, приводя к ПНЯ [34].
Ген PGRMC1 (мембранный компонент рецептора прогестерона 1) впервые был описан в 1998 г. [36]. Белок этого гена экспрессируется во многих тканях женского организма, включая печень, надпочечники, матку, обеспечивая передачу сигналов прогестерона [36–38]. PGRMC1 опосредует антиапоптотическое действие прогестерона на клетки гранулезы [38–40]. Mansouri M. et al. провели скрининг полиморфизма гена у 67 женщин с идиопатической формой ПНЯ, в ходе которого в 4,5% случаев был выявлен патогенный вариант гена (p.H165R), расположенный в домене цитохрома b5, который вызывает нарушение активации микросомального цитохрома P450, приводя к апоптозу фолликулов [38].
Ген FMR1 (Fragile mental retardation) локализован на длинном плече Х-хромосомы в локусе Хq27.8 [41, 42]. В 5 нетранслируемой области 1-го экзона данного гена содержатся тринуклеотидные CGG повторы [43].
Нормальное количество повторов CGG в гене FMR1 у представительниц российской популяции соответствует 28–36 [13]. Увеличение количества повторов от 54 до 199 известно как премутация гена [44]. При увеличении числа повторов более 200 формируется болезнь Мартина–Белла, которой подвержены только мальчики. Ген FMR1 обладает плейотропным эффектом, являясь наиболее частой и хорошо изученной причиной ПНЯ, при этом снижение тотального овариального резерва наблюдается не только в рамках премутации гена, но и при изменении числовых повторов как в сторону их увеличения, так и уменьшения по отношению к нормальному диапазону 28–36 [45, 46]. По международным данным, при спорадической форме ПНЯ премутация гена FMR1 встречается от 0,8 до 7,5% случаев, при семейной форме – до 13% случаев. В работе Шамиловой Н.Н. премутация выявлена в 3,1% случаев, в то время как при семейных формах – в 7,9% случаев, а при спорадических – в 1,1% (1/91) [13].
На основании представленного выше материала становится понятным, что данные гены играют одну из ключевых ролей на этапе раннего фолликулогенеза, предотвращая преждевременную лютеинизацию фолликулов, а также они отвечают за ранний стероидогенез и регуляцию процессов апоптоза в примордиальных фолликулах [13]. По данным некоторых авторов, в генезе несиндромальной формы ПНЯ участвуют гены, картированные на Х-хромосоме – BMP15, PGRMC1 и FMR1, в то время как при синдромальной форме – гены, преимущественно расположенные на аутосомах, – GDF9, FIGLA, FSHR, NOBOX, NR5A1, NANOS3, STAG3, SYCE, MCM8,9 [14, 15].
Ни одному из выше представленных генов не принадлежит ведущая роль в генезе развития ПНЯ, что можно объяснить вариабельностью размера выборок и разнообразием этнических групп в проведенных исследованиях.
Помимо наиболее перспективных генов-кандидатов, полиморфизм которых приводят к преждевременному истощению овариального пула, особый интерес представляет обсуждение роли генов PTEN и BRCA1,2, обладающих плейотропными эффектами.
Гены BRCA1 (17q21) и BRCA2 (13q12.3) относятся к группе генов-супрессоров, вовлеченных в процесс гомологичной репарации двунитевых разрывов ДНК. На сегодняшний день известно, что полиморфизмы в генах BRCA1 и BRCA2 отвечают за развитие наследственных форм рака молочной железы, яичников, маточных труб, простаты и поджелудочной железы [47–48]. Учитывая, что BRCA1 играет важную роль как в митотических, так и в мейотических процессах, физиологическое истощение ооцитов может протекать быстрее во внутриутробном периоде жизни плода в случае «поломки» гена BRCA1 [49, 50]. Ген BRCA1 экспрессируется в бластоцистах [51]. Концепция некоторых исследователей гласит, что ооциты с патологической функцией генов BRCA и последующим повреждением ДНК могут преждевременно гибнуть за счет ускоренного процесса апоптоза, что в свою очередь, приводит к раннему истощению фолликулярного овариального резерва [52, 53]. Другая теория состоит в том, что увеличение мутаций в примордиальных фолликулах препятствует нормальному созреванию ооцитов, что также впоследствии приводит к ускоренному их апоптозу [54]. Ряд исследований демонстрирует выраженное снижение уровня АМГ у носительниц патогенных вариантов гена BRCA1 в сравнении с контрольной группой [55, 56]. Известно, что при наличии полиморфизма гена BRCA1 в 30% случаев отмечено уменьшение числа СGG-повторов в гене FMR1 в сравнении с их нормативными показателями [57]. Существует несколько принципиально разноречивых мнений ученых относительно вклада данных генов в раннее истощение фолликулярного пула. В недавних исследованиях продемонстрировано, что патогенный вариант генов BRCA, особенно BRCA1, значительно уменьшают овариальный резерв яичников, что, в свою очередь, приводит к бесплодию [47, 52].
При обследовании 908 канадских женщин в возрасте от 18 до 87 лет, носительниц патологических вариантов генов BRCA1 и BRCA2, были выделены 253 пациентки старше 45 лет (1-я группа). Контрольную группу составили 908 женщин без нарушений в генах BRCA аналогичного возраста, при этом респондентов после 45 лет было 216 (2-я группа). Проведенное анкетирование показало, что ранняя менопауза в 1-й группе выявлена в 22,1% (56 женщин), а ПНЯ – в 4,7% (12 женщин), в то время как во 2-й группе эти показатели составили 14,3% (31 женщина) и 1,4% (3 женщины) соответственно. Таким образом, при наличии полиморфизма в генах BRCA1, -2 ранняя менопауза встречается в 1,5 раза чаще, ПНЯ – в 3,3 раза [53].
В доступной литературе нами не найдено исследований, в которых при ПНЯ анализировалась частота встречаемости патогенных вариантов BRCA1,2, в то время как в общей популяции среди европейских женщин это значение достигает 1 из 1000 случаев, однако в некоторых этнических группах эта цифра возрастает до 2,5% [58, 59].
Ген PTEN (гомолог фосфатазы и тензина), локализованный в локусе q23.3 хромосомы 10, играет одну из ведущих ролей в ранней активации первичных фолликулов, негативно регулируя AKT-сигнальный путь [60]. У мышей, нокаутных по гену PTEN, экспериментально доказано преждевременное истощение фолликулярного аппарата вследствие бесконтрольного рекрутинга фолликулов. В норме белок PTEN блокирует PI3K-АКТ путь, удерживая первичные фолликулы в фазе режима «покоя» [61].
Становится понятным, что гены-кандидаты чаще демонстрируют не только экспрессию в яичниках, но и на системном уровне. При этом NOBOX и FIGLA регулируют функциональное состояние только яичников, в то время как PTEN играет жизненно важную роль в активации первичных фолликулов, одновременно, являясь основным регулятором пути PI3K, регулируя процессы пролиферации, миграции и метаболизма организма на клеточном уровне в целом [16].
В последние годы доказано, что, помимо генов, принимающих участие в фолликуло- и стероидогенезе, на преждевременное снижение овариального резерва оказывают влияние гены, вовлеченные в процессы мейоза и репарации ДНК, выявляемые на основе NGS-методики [15]. В физиологических условиях внутриутробно ооциты в примордиальных фолликулах длительно замирают в стадии профазы I мейотического деления. Экспериментально доказано, что при отсутствии или патогенных вариантах генов, регулирующих механизмы мейотического деления, развивается ПНЯ [62, 63]. Гены STAG3 и SYCE1 необходимы для правильного формирования синаптонемного комплекса во время клеточного деления, и патогенные варианты этих генов приводят к бесплодию у людей и животных [63, 64]. Геликазы белков поддержания мини-хромосомы (MCM8 и MCM9) играют решающую роль на стадии гомологичной рекомбинации в процессе мейотического деления [65]. Гомозиготная мутация способствует развитию ПНЯ.
Таким образом, становится понятным, что ПНЯ – гетерогенное заболевание, и благодаря использованию NGS-технологий обнаружены различные дефекты в более чем 75 генах, участвующих в генезе преждевременного старения яичников [66]. В последние годы проводят полноэкзомное секвенирование (WES) геномной ДНК не только пациенток, но и их родителей с последующим совместным биоинформатическим анализом (семейные формы ПНЯ). В результате чего выявляется обширный список генетических отличий от нуклеотидной последовательности «референсного» генома, большая часть которых не имеет клинического значения. Проведение сравнения выявленных отличий у пациента и родителей позволяет обнаружить среди этого списка клинически значимые изменения в геноме пациента. Базы данных клинически значимых отклонений постоянно пополняются, и именно такие исследования во многом способствую этому процессу [67].
В настоящее время не существует достоверных биомаркеров для оценки овариального резерва. Ультразвуковой мониторинг и гормональный профиль (уровни ФСГ и АМГ в сыворотке крови) оценивают не тотальный, а функциональный резерв, то есть количество рекрутируемых в данном цикле примордиальных фолликулов [68].
Определенные надежды в плане предвидения ПНЯ возлагаются на определение miRNA (микросомальная рибонуклеиновая кислота) в сочетании с экзосомами, которые легко обнаруживаются в кровотоке, в связи с чем, этот метод в дальнейшем может быть использован в качестве биомаркера оценки овариального резерва и ранней диагностики ПНЯ [69]. Известно, что miRNA представляют собой семейство небольших некодирующих молекул РНК длиной 19–24 нуклеотидов, играющих важную регуляторную роль в экспрессии генов [69, 70]. Недавние открытия показывают, что miRNA являются важными регуляторами клеточной дифференцировки и вовлечены в процесс доимплантационного периода. Они поддерживают баланс между плюрипотентностью и дифференцировкой эмбриона и эмбриональных стволовых клеток. Снижение экспрессии miR-22-3p в плазме пациентов с ПНЯ коррелирует с объемом овариального пула. Авторы данного исследования также представили доказательство роли полиморфизма генов miR-518 и TGFBR2 в качестве новых предикторов риска развития ПНЯ и ранней менопаузы, а также возраста наступления естественного выключения функции яичников [71]. Взаимосвязь между miR-146 и miR-196a2 вовлечена в процесс формирования ПНЯ. Результаты микроматричного типирования продемонстрировали, что у пациенток с ПНЯ по сравнению с группой контроля 10 miRNA значительно активированы (miR202, miR146a, miR125b-2, miR139-3p, miR654-5p, miR27a, miR765, miR23a, miR342-3p, miR126), в то время как 2 miRNA (miR518, взаимодействие между miR146 и miR196a2) инактивированы. Среди активированных miRNAs обращает на себя внимание miR23a, играющая важную роль в процессе апоптоза в клетках гранулезы в яичниках посредством снижения экспрессии X-связанного ингибитора апоптоза белка (XIAP). Кроме того, обнаружено, что miR-22-3p демонстрирует более низкий уровень экспрессии у женщин с повышенным уровнем ФСГ по сравнению с группой контроля [69].
Заключение
Знание генов-кандидатов, лежащих в основе генеза формирования ПНЯ, и возможность в клинической практике проводить полноэкзомное секвенирование будут иметь первостепенное значение не только для понимания физиологии яичников, но и для генетического консультирования с целью оценки репродуктивного прогноза в группах риска по развитию ПНЯ. Женщинам-носительницам патогенных вариантов генов-кандидатов по ПНЯ на доклиническом этапе необходимо регулярно мониторировать гормональные показатели овариального резерва с целью своевременного забора их генетического материала с последующей криоконсервацией яйцеклеток.


