
По современным представлениям, тромбозы вен и артерий, тромбоэмболии, хронический синдром диссеминированного или локального внутрисосудистого свертывания крови (ДВС-синдром) являются грозными осложнениями у онкологических больных. Клинически венозный тромбоэмболизм и рак имеют два основных проявления: с одной стороны тромбоз может быть единственным клиническим симптомом скрыто протекающего рака, с другой – у пациентов с выявленным раком на всех стадиях заболевания может развиться тромбоз. В настоящее время уже не вызывает сомнений, что у больных онкологическими заболеваниями тромбозы и тромбоэмболии возникают значительно чаще, о чем свидетельствуют современные многоцентровые исследования.
Патогенез взаимодействия раковой клетки и системы гемостаза
В процессе опухолевой прогрессии маскировка злокачественно трансформированных клеток от эффекторов иммунной системы в числе прочего обеспечивается наличием барьера из фибрина. Фибриновые волокна всегда присутствуют в любой опухолевой ткани, что особенно характерно для зоны активной пролиферации малигнизированных клеток. В зоне опухоли мощный фибриновый барьер образуется за счет извращения процессов перманентной стимуляции фибриногенеза и сопутствующего фибринолиза. Со стороны опухолевых клеток инициирующее воздействие на компоненты системы гемостаза и процесс образования фибрина опосредуется присутствующими в зоне опухоли макрофагами, реакция которых на малигнизированные клетки выражается усилением продукции провоспалительных цитокинов: фактора некроза опухоли-α (TNF-α, ФНО-α), интерлейкинов (IL-1, IL-6, IL-8) и других активных молекул. На поверхности клеток эндотелия и активированных макрофагов провоспалительные цитокины индуцируют экспрессию тканевого фактора свертывания крови [1, 2].
В очаге опухолевого роста защитная роль фибрина искажается, и трансформированные клетки используют фибриновую сетку как механический барьер от клеток иммунной системы. Фибрин также повышает устойчивость малигнизированных клеток к химиотерапии. В целом, фибрин является для раковых клеток и основным трофическим материалом. Раковые клетки потребляют фибрин в 10 раз более активно, чем другие белки. В структуре опухолевой ткани фибрин выполняет, кроме того, функцию внеклеточного матрикса. Нити фибрина оказывают стимулирующее действие на фибробласты, присутствие которых в опухоли необходимо для поддержания концентрации потребляемых раковыми клетками ростовых факторов и секреции компонентов, входящих в строму опухолевой ткани. Злокачественно трансформированные клетки используют фибрин в своих целях благодаря их способности определенным образом контролировать активность фибринолитической компоненты гемостаза – системы плазмин – тканевые ингибиторы – активаторы плазминогена [3, 4].
Патогенез тромбофилии у онкологических пациентов включает факторы, связанные с ответом на опухоль (воспаление, острофазовая реакция, диспротеинемия, очаговые некрозы, гемодинамические нарушения), а также специфические факторы, обусловленные самими опухолевыми клетками и связанными c опухолью макрофагами. А именно: прокоагулянтная и фибринолитическая активность раковых клеток, их взаимодействие с тромбоцитами, мононуклеарными макрофагами и эндотелием, неоангиогенез, лечебные мероприятия (химиотерапия, гормонотерапия). Опухолевые клетки активируют коагуляционную систему или систему фибринолиза, создавая условия для дальнейшего своего распространения, стимуляции ангиогенеза, повышения сосудистой проницаемости, что в свою очередь способствует метастазированию.
Адгезия тромбоцитов к субэндотелиальным соединительнотканным компонентам осуществляется за счет взаимодействия гликопротеиновых рецепторов (GР) тромбоцитов с коллагеном субэндотелиального матрикса или с другими реактивными адгезивными протеинами, включающими фактор Виллебранда (vWF), фибриноген, фибронектин, ламинин, витронектин и тромбоспондин [5].
В процессе развития онкологического процесса определяющую роль играет взаимодействие между опухолевой тканью, системой гемостаза и иммунной системой. Иммунная система с одной стороны является системой, защищающей организм от внешних воздействий, с другой стороны препятствует распространению эндогенных патологических процессов. Однако в тесном взаимодействии с системой гемостаза иммунный ответ может, в свою очередь, спровоцировать развитие одного из самых грозных патологических процессов – тромбоза и тромбоэмболии.
Ключевым звеном развития воспаления является повреждение сосудистого эндотелия (рис. 1). Формирование эндотелиальной дисфункции происходит под действием широкого спектра медиаторов воспаления и обусловливает активацию процессов коагуляции, системы комплемента, клеток крови, в первую очередь макрофагов и нейтрофилов. В результате массивного выброса провоспалительных цитокинов происходит непрерывная стимуляция лейкоцитов, макрофагов, их неконтролируемая активация и высвобождение широкого спектра медиаторов воспаления, включая цитокины, фактор активации тромбоцитов (PAF), колониестимулирующие факторы, метаболиты цикла арахидоновой кислоты, тромбоксан А2, простагландины, лейкотриены, свободные радикалы кислорода, различные протеазы, которые участвуют в повреждении тканей [6].
В то же время лейкоциты могут доставлять тромбоциты в область воспаления, осуществляя механизм высвобождения тромбоцитарного фактора 4, тромбоцитарного фактора роста и других тромбоцитарных хемокинов. Адгезия активированных тромбоцитов к лейкоцитам повышает секрецию цитокинов или других медиаторов лейкоцита. Такие взаимодействия могут как усиливать воспаление, так и активировать систему гемостаза.
Кроме того, для злокачественных опухолей характерна инвазия в васкуляризированные ткани пораженного органа раковыми клетками и образование новых кровеносных сосудов, то есть опухолевый неоангиогенез. Также прокоагулянтные свойства раковых клеток реализуются посредством утечки факторов свертывания из плазмы в интерстициальное пространство микросреды опухоли, секрецией цитокинов, хемокинов и специфических опухолевых муцинов, а также прямой инвазией раковых клеток в кровеносные или лимфатические сосуды.
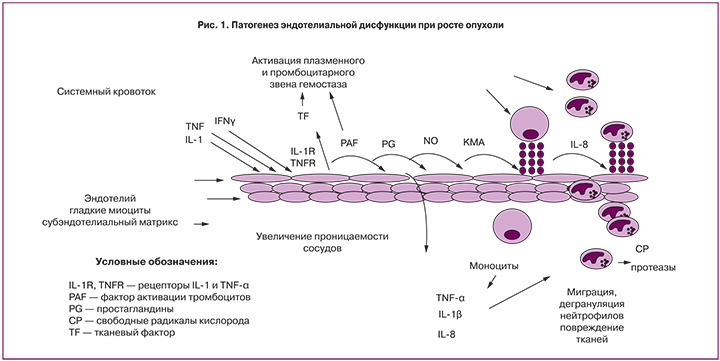
Повреждение стенки сосуда может быть обусловлено и циркулирующими воспалительными цитокинами, которые экспрессирует опухолевая ткань, такими как TNF-α или IL-1β и прямым действием цитотоксических лекарственных средств в результате эндотелиального повреждения. Например, при применении цисплатина происходит индукция апоптоза эндотелиальных клеток in vitro, в том числе за счет фрагментации мембраны; при этом микрочастицы обладают выраженной прокоагулянтной активностью, и увеличивается риск как венозных, так и артериальных тромбозов in vivo [7, 8].
Состояние гиперкоагуляции при раке обусловлено комплексом взаимодействий опухолевых клеток и их продуктов с клетками организма. Опухолевые клетки могут напрямую активировать коагуляционный каскад, что ведет к тромбозам, или проявлять прокоагулянтные свойства, или ингибировать антикоагулянтную систему эндотелия, тромбоцитов, моноцитов, макрофагов [4, 8]. Опухолевые массы вызывают по меньшей мере локальный стаз, обусловленный инвазией опухоли в кровеносные сосуды, механическим сдавлением массой опухоли.
Раковая клетка может инициировать коагуляцию непосредственно через взаимодействие с тромбоцитами и/или системами коагуляции и фибринолиза, чтобы генерировать тромбин, или косвенно, стимулируя мононуклеарные клетки, что ведет к синтезу различных прокоагулянтов. Запуск свертывания может рассматриваться как особый тип воспалительной реакции на стимулы типа повреждения стенки сосуда, или внутрисосудистой агрегации раковых клеток, или поступления клеток опухоли в кровоток. Увеличение прокоагулянтной активности, присутствие всех компонентов системы коагуляции локально в области расположения опухоли и уменьшение деятельности противосвертывающей системы ведет к гиперкоагуляции как результату злокачественного развития.
Генерация тромбина и формирование фибрина постоянно обнаруживается у онкологических пациентов, эти процессы приводят к увеличению риска тромбоэмболических осложнений. Более важно, что формирование фибрина вовлечено в процессы роста опухоли и метастазирования. Активация свертывания крови при раке – сложный феномен, включающий множество компонентов системы коагуляции и многочисленные взаимодействия между опухолевыми клетками и клетками крови, включая тромбоциты, моноциты, эндотелиальные клетки.
Значение тканевого фактора в патогенезе опухолевой активации коагуляции
Основными факторами, обусловливающими опухолевый ангиогенез, являются прежде всего тканевой фактор (ТФ, TF), тромбин, сосудистый эндотелиальный фактор роста (СЭФР), фибриноген и фибрин, а также продукты деградации фибрина. Стимулирующее влияние на процесс ангиогенеза в опухолевой ткани оказывают: ТФ, СЭФР, тромбин, фактор роста фибробластов (FGF-2), ТNF-a, интерлейкины, мембранные металлопротеиназы (MMPs), фибрин и его фрагменты. К факторам препятствующим опухолевому ангиогенезу относятся: тромбоспондин, эндостатин, интерфероны-a, b, g, антигепариновый фактор тромбоцитов (PF4), ангиопоэтин-1, фрагменты фибриногена [3, 7, 9].
Экспрессия ТФ в сочетании с действием цитокинов и различных факторов роста может изменять раковые клетки. ДНК раковых клеток изменяется таким образом, что образуются аберрантные формы ТФ, что обусловливает более злокачественный фенотип. ТФ играет ключевую роль как в физиологических условиях, так и при патологических формах ангиогенеза. ТФ и СЭФР участвуют в порочном цикле формирования тромба и роста опухоли. Не только ТФ стимулируют СЭФР, но и наоборот – СЭФР стимулирует экспрессию ТФ на эндотелиальных клетках [9, 10].
Крайне важно отметить, что некоторые виды опухолевых клеток способны самостоятельно синтезировать фибрин или фибриноген, однако большинство зависит от экстравазации фибриногена и формирования фибрина вокруг опухоли. В результате образования фибрина повышается проницаемость сосудов около очага опухолевого роста, фибриновая сеть или матрикс вокруг первичного очага роста способствует:
- Опухолевому росту первичного очага на основе фибринового каркаса;
- Защите от иммунной системы за счет образования «фибринового плаща»;
- Стимуляции экстравазации плазменных протеинов, что в первую очередь необходимо для опухолевого ангиогенеза;
- Усилению адгезии опухолевого эмбола, а точнее тромбоэмбола, к эндотелиальным клеткам вне области первичного очага, то есть метастазирования.
Патогенетическое значение тканевого фактора
Одним из важнейших компонентов опухолевой клетки, с помощью которого запускается активация внешнего пути свертывания, является ТФ. Он экспрессируется на поверхности мембраны [1], что в контексте опухолевого ангиогенеза означает повышение проницаемости, и как результат все компоненты, присутствующие в плазме, непосредственно проникают в интерстициальную среду и могут взаимодействовать с клетками опухоли. Также ТФ может повышать активность циклооксигеназы-2 (ЦОГ-2) и продукцию активатора ингибитора плазминогена 1-го типа (PAI-1), что в свою очередь является дополнительным фактором гиперкоагуляции [7, 9].
Иммунный ответ и процесс коагуляции могут взаимно активировать друг друга, тем самым приводя к формированию порочного круга. В частности, два из основных цитокиновых медиаторов воспаления – IL-1β и TNF-α обладают прокоагулянтным эффектом in vivo и вызывают экспрессию TФ, а также снижают уровень антикоагулянта тромбомодулина на поверхности клеток in vitro. Кроме того, помимо функции инициатора процессов коагуляции, ТФ обладает еще и провоспалительной активностью. Одновременно с этим, повреждение ткани запускает процесс активации коагуляции – тромбин и фактор Xa, взаимодействуя с рецепторами из семейства рецепторов активированных протеаз (PARs), дополнительно активируют продукцию провоспалительных цитокинов [7, 10, 11]. При этом ТФ занимает ключевую позицию во взаимодействии опухолевой ткани, системы гемостаза и иммунной системы (рис. 2).
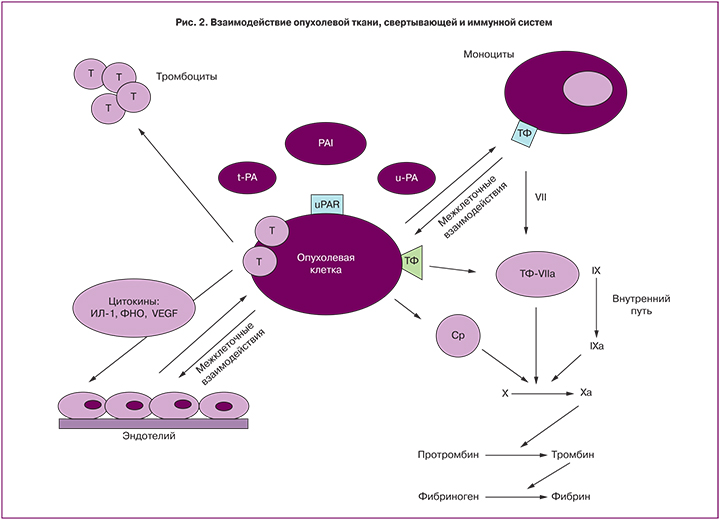
Опухолевые клетки экспрессируют: а) раковые прокоагулянты, активирующие каскад свертывания: ТФ, цистеиновая протеаза (CP); б) активаторы и ингибиторы фибринолиза, а также их рецепторы: урокиназный активатор плазминогена (u-PA), тканевой активатор плазминогена (t-PA), PAI, рецепторы урокиназного активатора плазминогена (u-PAR); в) цитокины, взаимодействующие с эндотелием путем его повреждения, активируют прокоагулянтные свойства эндотелиоцитов: IL-1, TNF, СЭФР. Кроме того, опухолевые клетки напрямую взаимодействуют с клетками крови: тромбоцитами, эндотелием, моноцитами [10, 12].
Большое значение имеет сочетание с генетическими формами тромбофилии и циркуляцией антифосфолипидных антител, чаще всего выявляются: мутация фактора свертывания V (FV) Leiden, мутация метилентетрагидрофолатредуктазы (MTHFR C677T), полиморфизм PAI-1 G4/G5, полиморфизм тромбоцитарных гликопротеинов, дефицит антитромбина III (AT III). На настоящий момент недостаточно данных как о частоте генетических форм тромбофилии у онкологических больных, так и клинических проявлениях при их наличии. Из наших наблюдений можно сделать вывод, что наличие генетической тромбофилии, особенно мутации MTHFR, может быть серьезным фактором, провоцирующим клинически выраженные тромботические осложнения, а особенно скрытые тромбозы, которые выявляются как патологоанатомические находки в 3–4 раза чаще [13]. Вне всякого сомнения, при наличии генетических форм тромбофилии патогенез тромботических осложнений обусловлен в основном истощением противосвертывающей системы. На основании данных других авторов и собственных наблюдений мы можем рекомендовать при наличии мутации MTHFR включение в терапевтическую схему фолиевой кислоты и витаминов группы В. А также при наличии мультигенных форм тромбофилии мы можем рекомендовать перманентную антикоагулянтную терапию под контролем маркеров тромбофилии: D-димера, комплекса тромбин-антитромбин (ТАТ) и PF4.
Кроме этого у различных опухолевых клеток присутствует еще ряд факторов, также оказывающих влияние на активацию системы гемостаза. В частности это могут быть онкогены, такие как протоонкоген, представитель семейства белков Ras (KRAS), который активирует белки, необходимые для распространения факторов роста. Например, ген KRAS часто обнаруживают при лейкемиях, раке толстой кишки, раке поджелудочной железы и раке легких. Определение мутационного статуса гена KRAS имеет значение при планировании терапии препаратами блокаторами эндотелиального фактора роста (EGFR), так как карциномы с мутантным KRAS резистентны к терапии анти-EGFR моноклональными антителами. Также значение может иметь и ген рецептора ретиноевой кислоты типа α (RARα) и ген белка острого промиелоцитарного лейкоза (PML), что приводит к образованию аномального онкопротеина PML-RARα, что в свою очередь ведет к неконтролируемому размножению мутантных промиелоцитов. Наличие подобных проонкогенов дополнительно может усиливать гипоксию и провоспалительную реакцию в микроокружении опухоли и являться дополнительным фактором образования фибрина и опухолевого неоангиогенеза [5].
Адгезия тромбоцитов к субэндотелиальным соединительнотканным компонентам осуществляется за счет взаимодействия гликопротеиновых рецепторов тромбоцитов с коллагеном субэндотелиального матрикса или с другими реактивными адгезивными протеинами, включающими vWF, фибриноген, фибронектин, ламинин, витронектин и тромбоспондин [14].
Тромбин, кроме того что участвует в активации коагуляции, играет существенную роль в росте опухоли и метастазировании, увеличивая адгезивные свойства опухолевых клеток, и действует как проангиогенный фактор. Также тромбин стимулирует PAI ткани, ингибируя, фибринолитическую систему [6, 12, 15].
И гемостаз-зависимые, и гемостаз-независимые механизмы вызывают ангиогенез. ТФ индуцирует гемостаз-независимый механизм через фосфорилирование эндоплазматического хвоста и последующих каскадов трансдукции сигнала. ТФ индуцирует ангиогенез через гемостаз-зависимый механизм, генерируя тромбин. Тромбин также стимулирует ангиогенез через гемостаз-независимые и зависимые механизмы. Гемостаз-независимые механизмы опосредуются путем расщепления PARs и последующей активации каскада трансдукции сигнала связанной с протеином G, которые стимулируют связанные с ангиогенезом гены. Гемостаз-зависимые механизмы опосредуются через осаждение фибрина и активацию тромбоцитов [5, 8].
СЭФР и TNF-α также регулируют прокоагулянтную активность ТФ, что ведет к развитию системной гиперкоагуляции, свойственной многим раковым пациентам.
Таким образом, тромбофилическое состояние и ДВС-синдром, развивающиеся при наличии новообразования, имеют сложный многокомпонентный патогенез. Однако принципиально важным является тот факт, что тромбофилическое состояние – не просто сопутствующий злокачественному новообразованию процесс, как, например, сдавление окружающих органов опухолевыми массами, обусловливающее ишемию этих органов или интоксикация организма при развитии опухолевого процесса. При активации системы гемостаза, приводящей к развитию ДВС-синдрома, создаются условия для роста опухолевой ткани за счет опухолевого ангиогенеза и распространения за счет метастазирования раковых клеток. Таким образом, контроль за состоянием системы гемостаза и предупреждение тромбофилии является не только профилактикой тромбогеморрагических осложнений у онкологических больных, но и лечением основного заболевания за счет блокирования путей роста (ангиогенеза) и метастазирования опухоли [3, 16].
Процессы опухолевого роста и развития тесно связаны между собой, соответственно не менее тесная связь существует и с процессом воспаления. Активация свертывающей системы крови в ответ на воспаление, обусловленное наличием опухоли, служит защитным механизмом, направленным на ограничение участка повреждения тканей и предупреждение дальнейшего распространения патогенного фактора в организме. В случае системного воспаления этот механизм теряет свое адаптивное значение. В настоящее время под системным воспалительным ответом организма понимают нарушение гомеостаза между процессами воспаления, коагуляции и фибринолиза [8, 14].
Общая патология опухолевого неоангиогенеза
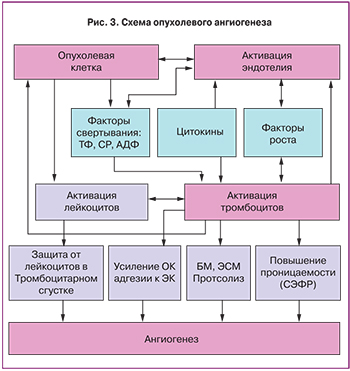 В основе патогенеза гемостазиологической паранеоплазии лежит активация как коагуляционного, так и сосудисто-тромбоцитарного звеньев свертывания крови, что обеспечивается:
В основе патогенеза гемостазиологической паранеоплазии лежит активация как коагуляционного, так и сосудисто-тромбоцитарного звеньев свертывания крови, что обеспечивается:
- нарушением структурной целостности и функциональной стабильности сосудистого эндотелия опухолевыми клетками и цитокинами;
- активацией тромбоцитов опухолевыми клетками, приводящей к их повышенной адгезии и агрегации;
- синтезом прокоагулянтов и ингибиторов фибринолиза опухолевыми клетками;
- прокоагулянтной активностью опухоль-ассоциированных макрофагов и активированных моноцитов периферической крови.
Схема опухолевого ангиогенеза представлена на рис. 3.
Ангиогенез требуется для роста и метастазирования злокачественных опухолей. Ангиогенез управляется разнообразными факторами, включая СЭФР и антиангиогенный тромбоспондин. Была обнаружена связь между уровнем ТФ и ангиогенной активностью, а также с уровнем СЭФР [17].
Многочисленные доклинические и клинические исследования показали зависимость роста опухоли от процесса ангиогенеза. Активация коагуляционного каскада зачастую обнаруживается у пациентов с раковыми опухолями. Тромбоциты принимают участие в опухоль-индуцированном ангиогенезе. Это утверждение основано на фактах, что тромбоциты богаты стимуляторами и ингибиторами ангиогенеза и взаимодействуют с эндотелием. Антитромботическое состояние эндотелия нарушается стимулами, исходящими от опухолевых клеток [18].
С точки зрения биологии опухоли, местные явления в пределах опухоли и на ее поверхности, а также внешних кровеносных сосудах, имеют главное значение. Формирование фибрина вызвано ТФ, который активирует коагуляционный каскад, активируя фактор X,
преобразование протромбина в тромбин и затем фибриногена в фибрин и стабилизацию фибрина. Фибрин – механическая поддержка для опухоли, способствует ее росту. Осаждения фибрина на поверхности опухоли является барьером для иммунной системы. Фибрин облегчает ангиогенез. Иммунохимический анализ показал наличие обильной сети капилляров на местах, богатых отложениями фибрина. Фибриноген создает условия для метастазирования. Фибриновая сеть представляет собой временный матрикс, который обеспечивает реваскуляризацию поврежденной ткани. Образование фибрина является результатом активации свертывания крови, которая сопровождает такие процессы, как тромбоз, рост опухоли, воспаление и играет при этом существенную роль в патологическом ответе тканей. Вслед за повреждением тканей происходит экстравазация фибриногена из кровеносных сосудов в экстраваскулярное пространство с образованием фибринового матрикса. Воспалительные и эндотелиальные клетки мигрируют в такой матрикс и стимулируют процессы репарации. Отложения фибрина вокруг опухоли имеют определенные функции. Главным образом осаждение фибрина является основой формирования начальной структуры опухоли. Через какое-то время гель фибрина преобразовывается в зрелую васкуляризированную соединительную ткань путем активации ангиогенеза. Фибрин имеет существенное влияние на воспалительную инфильтрацию опухоли, заключающееся в регулировании формирования стромы и защиты опухоли от иммунной системы [5, 11, 18].
Адгезия тромбоцитов к субэндотелиальным соединительнотканным компонентам осуществляется за счет взаимодействия GР тромбоцитов с коллагеном субэндотелиального матрикса или с другими реактивными адгезивными протеинами, включающими vWF, фибриноген, фибронектин, ламинин, витронектин и тромбоспондин [19].
Рецептором фибриногена на тромбоците является интегрин aIIbb3. Для взаимодействия с фибриногеном тромбоциты должны быть активированы тромбином, аденозиндифосфатом (АДФ), адреналином или коллагеном. Эти вещества выбрасываются в значительных количествах при повреждении сосудистой стенки и, связываясь со специфическими рецепторами на поверхности тромбоцитов, запускают каскад внутриклеточных активаторов. Активирующие влияния приводят к экспрессии рецептора фибриногена и изменению его конформации, что ведет к активации рецептора [20].
Тромбин, кроме того что участвует в активации коагуляции, играет существенную роль в росте опухоли и метастазировании, увеличивая адгезивные свойства опухолевых клеток, и действует как проангиогенный фактор. Рецепторы с высокой аффинностью к тромбину были обнаружены на поверхности опухолевых клеток. Также тромбин стимулирует PAI ткани, ингибируя фибринолитическую систему. Не все опухоли характеризуются таким типом местного взаимодействия с системой коагуляции. Существует множество типов опухолей, не выделяющих ТФ и не вызывающих образование тромбина, но экспрессирующих активаторы плазмогена t-PA, особенно урокиназный u-PA. Деградация внеклеточных матричных белков облегчена чрезмерной экспрессией u-PA, t-PA и u-PAR. t-РА является ферментом эндотелиальной клетки. К факторам, регулирующим его секрецию эндотелием, относятся тромбин, гистамин, ацетилхолин, брадикинин, адреналин и интерлейкины. В плазме t-РА циркулирует в виде комплекса с естественным РАI-1, и лишь 5% и меньше t-РА циркулирует в свободной активной форме. Другим важным активатором плазминогена является активатор u-РА [9, 14, 21].
Значение эндотелиальной активации в патогенезе опухолевого роста
Сосудистые эндотелиальные клетки играют главную роль во всех механизмах, которые способствуют воспалительно-индуцированной активации коагуляции. Во время острой инфекции эндотелий активируется патогенами или посредством медиаторов воспаления. Провоспалительные цитокины, включая IL1, TNFα, индуцируют ТФ в эндотелиальных клетках, который частично «теряется» в растворимой форме. Этот «потерянный» растворимый ТФ и объясняет всю сложность определения эндотелиального ТФ и иммуногистохимических исследования на животных. Эти провоспалительные цитокины вызывают угнетение регуляции антикоагулянтных рецепторов ТФ и эндотелиальный рецептор протеина C (EPCR). Растворимый ТФ может также играть и роль ко-фактора при активации тромбин-активированного фибринолитического ингибитора (ТАФИ), который медленно угнетает фибринолиз и ведет к ДВС. Применительно к воспалению можно отметить, что уменьшение ТФ и EPCR на поверхностях клеток, как это было продемонстрировано на примере микрососудов больных сепсисом, ведет к увеличению воспалительного ответа in vivo [7, 17].
ТАФИ синтезируется и выделяется эндотелиальными клетками. Однако нет данных за то, что недостаток или дисфункция ТАФИ имеет место при инфекционных болезнях, таких, например, как сепсис, а также не доказана его клиническая роль в воспалении и инфекции.
Исследования доказали, что система протеина С крайне важна при воспалении и инфекции. Тромбин катализирует активацию протеина С, что модифицирует воспаление, апоптоз и может привести к излечиванию, хотя точный механизм противовоспалительного действия до конца не выяснен. EPCR, возможно, тоже играет роль в воспалении, но детально его участие до конца не изучено [22].
Другие важнейшие эндотелиальные клеточные механизмы действуют посредством PARs. Эти рецепторы широко распространены среди различных клеточных типов. Они соединяются с клеточными сигнальными факторами, которые регулируют эндотелиально-зависимую релаксацию и контракцию, ангиогенез, сосудистую проницаемость и экспрессию Р-селектина, межклеточных адгезивных молекул (МАМ) и сосудистых клеточных адгезивных молекул (СКАМ). Фосфорилирование клеточных протеинов имеет обратную зависимость по отношению к экспрессии рецепторов, что регулирует уровень активности. Тромбин, трипсин и активированный протеин С (АРС) являются агонистами PAR-1; PAR-2 активируется главным образом факторами Xа и VIIа. Недавнее исследование кинетики PAR-1 показало, что АРС действительно может активировать PAR, но примерно в 104 раза слабее, нежели тромбин, и необходимая концентрация намного выше, чем таковая при эндогенном АРС in vivo. Таким образом, тромбин считается главным агонистом PAR-1 на эндотелиальных клетках. Активаторы коагуляционной системы, в особенности тромбин, могут стимулировать эндотелиальные клетки на экспрессию провоспалительных цитокинов, таких, как IL-6, IL-8 и моноцитный гемотактический протеин-1 (МНП-1) [23].
Эндотелиальные клетки также экспрессируют адгезивные молекулы и фактор роста, что не только существенно для возникновения воспалительного ответа, но и увеличивает коагуляционный ответ. Взаимодействие между тромбоцитами и эндотелиальными клетками, как и между тромбоцитами и нейтрофилами, важно при воспалении. В эндотелиальных клетках Weibel-Palade тельца секретируют фактор vWF и Р-селектин, которые поддерживают функцию тромбоцитов. Активированные тромбоциты совместно с лейкоцитами и эндотелиальными клетками поддерживают концентрацию медиаторов при воспалительном ответе. Таковыми медиаторами являются CD 40 – лиганд, липоксигеназы, простагландины и др. [6, 14]
Система фибринолиза играет важную роль в патогенезе воспаления. У мышей с дефицитом активаторов плазминогена при введении эндотоксина наблюдается усиление процессов отложения фибриновых сгустков в органах и тканях по сравнению с контрольной группой, тогда как у мышей с дефектом гена PAI-1 при введении эндотоксина тромбозы микроциркуляторного русла не возникают.
Важно отметить, что активации коагуляции предшествует быстрая транзиторная активация фибринолиза. Внутривенное введение эндотоксина вызывает быстрый ответ системы фибринолиза. Сначала происходит высвобождение активатора фибринолиза тканевого и урокиназного типа, а затем следует быстрый подъем уровня PAI-1 за счет усиления его экспрессии под действием провоспалительных цитокинов (преимущественно TNF-α и IL-1β). Высокий уровень PAI-1 является прогностически неблагоприятным признаком у пациентов с ДВС-синдромом. Через 2 ч после введения эндотоксина происходит активация коагуляции. Увеличенное образование тромбина определяется по нарастанию в плазме концентрации пептидов фрагментов протромбина 1+2 (F1+2), образующихся при активации протромбина, а также ТАТ. В то же время уже через 3 ч после введения эндотоксина ресурсы фибринолитической системы оказываются исчерпанными, и конечным результатом эндотоксинемии является прокоагулянтное состояние [18, 24].
Фибриноген и фибрин, в свою очередь, способствуют формированию воспалительного ответа. Эти факторы непосредственно стимулируют экспрессию провоспалительных цитокинов TNF-α и IL-1β на мононуклеарных клетках и активируют продукцию хемокинов (IL-8, протеина хемотаксиса моноцитов (МСР-1)) эндотелиальными клетками и фибробластами. Считается, что эффекты фибриногена на мононуклеарные фагоциты опосредованы его взаимодействием с рецептором TLR-4, который одновременно служит рецептором эндотоксина. Под действием фибрина активируется адгезия лейкоцитов к эндотелиальным клеткам вследствие усиления экспрессии на их поверхности молекул клеточной адгезии 1-го типа (ICAM-1). Кроме того, связываясь одновременно с ICAM-1 на поверхности эндотелия и с активированными лейкоцитами, он формирует своего рода мост, поддерживающий адгезию [25].
Компоненты фибринолитической системы, в частности u-РА и его рецептор u-PAR, принимают участие в регуляции воспалительного ответа, что связано с их способностью модулировать процессы клеточной миграции. u-PAR активирует адгезию лейкоцитов к сосудистой стенке или к компонентам субэндотелиального матрикса, например к витронектину. Показано, что миграционная и инвазионная способность лейкоцитов коррелирует с уровнем экспрессии u-PAR на их поверхности [7, 14, 22]. Усиление экспрессии u-PAR на активированных мононуклеарах играет роль в их привлечении в зону ишемии у пациентов с инфарктом миокарда. В качестве одного из возможных механизмов активации миграции лейкоцитов называют усиление деградации экстрацеллюлярного матрикса под действием протеаз (эластазы, плазмина, металлопротеиназ), продукция которых регулируется при взаимодействии u-РА с его рецептором. Кроме того, при связывании u-PAR с витронектином активируется трансмембранный сигнальный путь, приводящий к синтезу цитокинов и факторов роста [9, 17].
В недавних исследованиях обнаружена противовоспалительная активность PAI-1. Последний, связывая витронектин, препятствует взаимодействию интегринов лейкоцитов с экстрацеллюлярным матриксом, таким образом, предотвращая их адгезию и миграцию. К тому же PAI-1 конкурирует с u-PAR за связывание с витронектином, тем самым уменьшая способность последнего активировать адгезию и миграцию лейкоцитов. In vitro обнаружена способность PAI-1 ингибировать эндотоксин-индуцированную продукцию TNF-α мононуклеарами. In vitro плазмин усиливает продукцию провоспалительных медиаторов моноцитами за счет активации митоген-зависимой протеинкиназы [15].
Вышеописанные патофизиологические особенности делают рак, возможно, наилучшим примером приобретенной тромбофилии. Улучшение понимания патофизиологии тромбофилии при раке должно обеспечить клиницистов более точным обоснованием более агрессивных и определенных стратегий противотромботического лечения у больных злокачественными опухолями.


