

Diagnosis of Noonan syndrome in pregnant women with nonimmune hydrops fetalis: tactics, outcomes, counseling
Lyushnina D.G., Shubina Je., Tetruashvili N.K., Zaretskaya N.V., Bolshakova A.S., Pak V.S., Kuznetsova M.V., Mikhailovskaya G.V., Bokeriya E.L., Trofimov D.Yu.
Relevance: Conducting extended genetic testing based on whole exome sequencing allows for the diagnosis of Noonan syndrome in pregnant women with nonimmune hydrops fetalis (NIHF). The data obtained may help to revise the management strategy for the current pregnancy and assist in determining the risk of recurrence
of Noonan syndrome in this parental couple.
Materials and methods: Pregnant women with NIHF underwent invasive prenatal diagnostics, followed by genetic testing using molecular karyotyping on DNA microarrays and whole exome sequencing.
Results: The study presents the clinical observations of three cases of NIHF associated with Noonan syndrome in the fetus. Married couples were examined and the inheritance pattern of variants associated with Noonan syndrome was determined. Genetic counseling was provided to the families, and a method for planning subsequent pregnancies was established.
Conclusion: Whole exome sequencing enables the diagnosis of Noonan syndrome in patients with NIHF, provides an estimation of fetal prognosis, and expands the scope of genetic counseling for the couple.
Authors' contributions: Lyushnina D.G., Shubina Je., Zaretskaya N.V., Tetruashvili N.K., Bokeriya E.L., Trofimov D.Yu. – conception and design of the study; Lyushnina D.G., Pak V.S., Bolshakova A.S., Mikhailovskaya G.V., Kuznetsova M.V. – material collection and processing; Lyushnina D.G., Shubina Je. – statistical analysis; Lyushnina D.G., Shubina Je., Tetruashvili N.K. – drafting of the manuscript; Tetruashvili N.K., Bolshakova A.S., Shubina Je., Bokeriya E.L., Trofimov D.Yu. – editing of the manuscript.
Conflicts of interest: The authors have no conflicts of interest to declare.
Funding: State assignment on the topic: "Development of a test system for prenatal diagnostics of fetal cardiopathology".
Ethical Approval: The study was reviewed and approved by the Research Ethics Committee of the V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia.
Patient Consent for Publication: All patients provided informed consent for the publication of their data.
Authors' Data Sharing Statement: The data supporting the findings of this study are available upon request from the corresponding author after approval from the principal investigator.
For citation: Lyushnina D.G., Shubina Je., Tetruashvili N.K., Zaretskaya N.V., Bolshakova A.S., Pak V.S., Kuznetsova M.V., Mikhailovskaya G.V., Bokeriya E.L., Trofimov D.Yu. Diagnosis of Noonan syndrome
in pregnant women with nonimmune hydrops fetalis: tactics, outcomes, counseling.
Akusherstvo i Ginekologiya/Obstetrics and Gynecology. 2024; (10): 34-40 (in Russian)
https://dx.doi.org/10.18565/aig.2024.196
Keywords
pregnancy
nonimmune hydrops fetalis
Noonan syndrome
RASopathy
RIT1
PTPN11
LZTR1
whole-exome sequencing
ascites
generalized edema
cystic hygroma
polyhydramnios
Noonan syndrome is a hereditary disorder that exhibits an autosomal dominant inheritance pattern. It affects multiple organs and systems with a phenotype that varies in severity, resulting in a diverse clinical picture [1, 2]. Estimates suggest that the prevalence of Noonan syndrome ranges from 1:1000 to 1:2500 live births [2–4]. However, the disease is often diagnosed only in late childhood owing to its delayed onset and progression. Prenatal diagnosis of Noonan syndrome is challenging because clinical manifestations at the antenatal stage are nonspecific. Additionally, the lack of mandatory genetic testing for the fetus or child in cases of perinatal loss makes it difficult to accurately determine the frequency of Noonan syndrome. The syndrome is characterized by a distinctive phenotypic triad that includes craniofacial dysmorphisms, congenital heart defects, and short stature. Clinical presentation can range from mild phenotypic features in adults without serious medical issues to severe manifestations observed antenatally, such as heart defects, skeletal abnormalities, and non-immune hydrops fetalis (NIHF) [2].
Noonan syndrome is a genetic disease associated with RAS pathologies, resulting from pathogenic variants in several genes, including PTPN11 (12q24.13), SOS1 (2p22.1), RAF1 (3p25.2), RIT1 (1q22), KRAS (12p12.1), BRAF (7q34), and LZTR1 (22q11.21), with rare variants found in other genes linked to the RAS/MAPK signaling pathway. This genetic heterogeneity partly accounts for the observed phenotypic variability among the patients. For instance, variants in PTPN11 and SOS1 are more commonly associated with pulmonary valve stenosis, whereas variants in RAF1 and RIT1 are associated with hypertrophic cardiomyopathy [5–7].
Fetuses with Noonan syndrome often exhibit prenatal features related to the NIHF symptom complex, including increased nuchal translucency, edema of the subcutaneous tissue, neck cystic hygroma, pleural or pericardial effusion, ascites, polyhydramnios, congenital heart defects, impaired kidney formation, and specific dysmorphic facial features [8, 9].
In our country, molecular genetic testing for NIHF is not regulated, leading to a frequent lack of known etiologies, including RASopathies. In some cases, when a normal karyotype is diagnosed in a fetus with NIHF and pregnancy is prolonged, dynamic observation is conducted. However, karyotyping does not rule out monogenic diseases, which necessitate whole exome sequencing of such fetuses. Whole-exome sequencing performed during pregnancy can assist in establishing a diagnosis of NIHF of unclear etiology.
Noonan syndrome accounts for 24–33.3% of NIHF in fetuses with a normal karyotype. Not all cases of the disease at the antenatal stage exhibit the phenotypic changes associated with Noonan syndrome, which complicates prenatal diagnosis [2, 3, 10, 11]. Typically, prenatal diagnosis of Noonan syndrome is performed only by testing couples with children with developmental defects linked to this syndrome [2]. This paper presents three clinical cases of NIHF associated with Noonan syndrome in fetuses.
Materials and methods
Pregnant women with NIHF were examined at the 2nd Obstetric Department of Pregnancy Pathology and the Institute of Reproductive Genetics of V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, from October 2021 to June 2024. All patients underwent transabdominal amniocentesis and amniotic fluid analysis to isolate the DNA for subsequent karyotyping and fetal exome sequencing. Whole-exome sequencing was performed if the fetus had a normal karyotype. Peripheral blood samples were collected from the parental couple, and written informed consent was obtained for whole-exome trio sequencing. Molecular karyotyping of the DNA microarrays (CMA) was performed to determine the karyotype. CMA was performed using the GenoScan3000 system on CytoScan Optima microarrays (Thermo Fisher Scientific, USA) according to the manufacturer's protocol. DNA was isolated from blood cells using the QIAamp kit. DNA sequencing was performed using a NovaSeq 6000 platform (WES, whole exome sequencing). The IDT XGen Exome Hyb Panel v2 enrichment kit was used to enrich target fragments. Sequencing data were processed using an algorithm that included read alignment to the hg38 reference genome sequence, calling, and filtering of variants by quality. All variants that passed quality filtering were annotated using the Ensembl Variant Effect Predictor (VEP) and several variant significance prediction algorithms (SIFT, PolyPhen-2, and SpliceAI). The Genome Aggregation Database (gnomAD) was used to estimate the population frequencies of the identified variants. To assess the clinical relevance of the identified variants, OMIM, ClinVar, LOVD, and other specialized databases (when available) as well as literature data were utilized. The clinical significance (pathogenicity) of the identified variants was assessed based on the ACMG recommendations and Russian guidelines for interpreting data obtained through high-throughput sequencing. The conclusion includes variants that may be related to the clinical manifestations in the patient as well as incidental findings, according to the list of genes for reporting secondary findings from ACMG.
Results
During invasive fetal diagnosis performed on pregnant women with NIHF, the fetuses were found to have Noonan syndrome in three cases. Table 1 illustrates the genes involved and inheritance patterns of Noonan syndrome among the examined pregnant women.

Clinical case 1
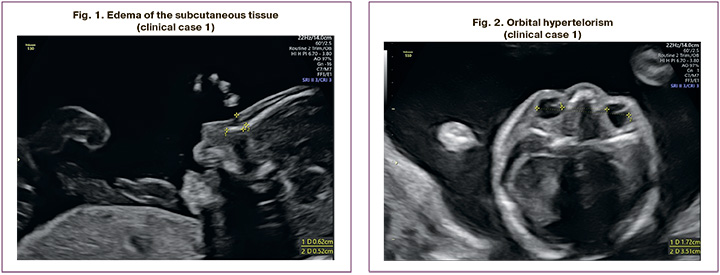
The patient, M.N., was a 28-year-old woman, who presented at 21 weeks of her second pregnancy. Ultrasound imaging showed swelling of the subcutaneous tissue of the neck and upper half of the fetal body (Fig. 1), bilateral hydrothorax, hypertelorism (Fig. 2), bilateral pyelectasis, fetal ventricular septal defect (VSD), hydropericardium, and polyhydramnios. In light of the perinatal period of 22–23 weeks, ongoing pregnancy, and established normal karyotype in the fetus, a decision was made to perform whole exome sequencing in the "trio" format. At 26 weeks, premature rupture of the membranes and spontaneous labor occurred. A live, extremely premature boy was born, weighing 1170 grams and measuring 36 cm. His Apgar score was 2–3. The newborn exhibited general edema syndrome, bilateral hydrothorax, intermuscular VSD, atrial septal defect (ASD), aortic isthmus hypoplasia, and ventricular hypertrophy.
On the second day of the child's life, the result of whole-exome sequencing was obtained. It reported a pathogenic heterozygous variant of the nucleotide sequence in the RIT1 gene, described in patients with Noonan syndrome. This variant was not detected in the parents, indicating that it arose de novo. The identified variant was validated by Sanger sequencing. The results of the molecular genetic study enabled the diagnosis of Noonan syndrome to be established. The child died on the 14th day of life.
Clinical case 2
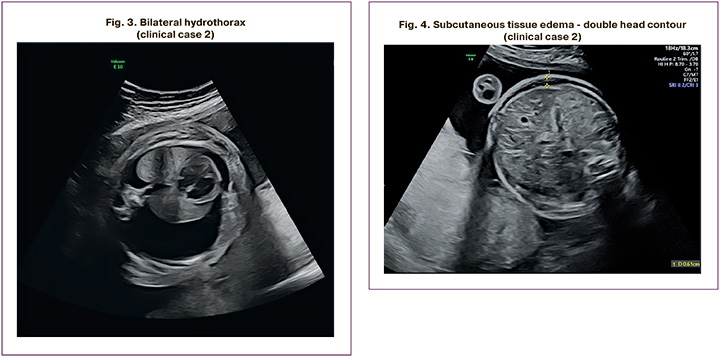
The patient, P.I., was a 37-year-old woman who presented at 37 weeks of her first pregnancy. She was diagnosed with NIHF, including left-sided hydrothorax and neck cystic hygroma of the fetal neck. At 24 weeks, there was bilateral hydrothorax (Fig. 3), neck cystic hygroma, generalized edema (Fig. 4), ascites, polyhydramnios, and placental cyst. At 26 weeks, the patient was referred to the Center for invasive prenatal diagnostics, which yielded a normal molecular karyotype. At 29 weeks, the fetus died antenatally. Autopsy revealed the presence of bilateral hydrothorax and generalized edema. Whole exome sequencing revealed a pathogenic heterozygous variant in the PTPN11 gene associated with Noonan syndrome, which had arisen de novo in the fetus. The identified variant was validated using Sanger sequencing.
Clinical case 3
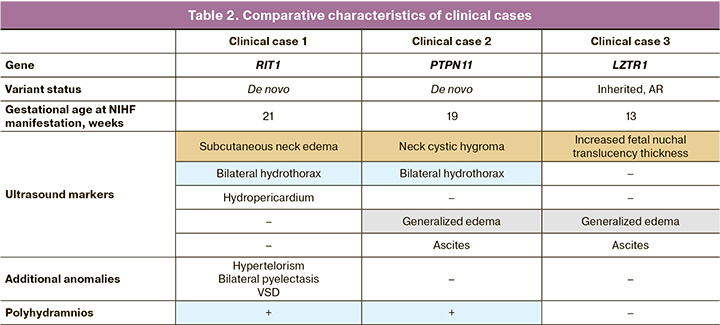
The patient, R.E., was a 32-year-old woman who presented with a second pregnancy. The first pregnancy resulted in a healthy child. Prenatal screening in the first trimester at 13 weeks revealed increased fetal nuchal translucency thickness, generalized edema, and ascites. At 14 weeks, invasive prenatal diagnostics were performed; however, given the extremely unfavorable prognosis for the fetus, the patient decided to terminate the pregnancy at her place of residence. The fetus was found to have two heterozygous variants in the LZTR1 gene inherited from the mother and father. The variants have been described in compound heterozygous forms in patients with Noonan syndrome. As part of the study, a conditionally healthy child from the family was examined, in whom one heterozygous variant was identified.
In 2024, the patient had her third pregnancy, during which the fetus was visualized at 9 weeks, with subcutaneous fat edema of the cephalic end and trunk. At 10 weeks of pregnancy, chorionic villus biopsy was performed, and frequent aneuploidies were excluded from the fetus. Sanger sequencing again identified variants in the LZTR1 gene. Given the presence of Noonan syndrome in the fetus and an extremely unfavorable prognosis, the pregnancy was terminated at 10 weeks for medical reasons. The comparative characteristics of these clinical cases are presented in Table 2.
The data presented reflect the non-specificity and heterogeneity of the phenotypic manifestations of Noonan syndrome in the prenatal period. However, pathological changes in the cervical region of the fetus were confirmed in all cases. As shown in the presented data, in only one clinical case, the typical structural abnormalities described in Noonan syndrome were found, that is, craniofacial dysmorphisms and congenital heart defect.
Discussion
Pregnant women with NIHF and a normal fetal karyotype are at risk for Noonan syndrome, which necessitates extended genetic testing such as whole-exome sequencing. According to global literature, the incidence of RASopathies among fetuses with NIHF is 30%, with Noonan syndrome accounting for 68% of these cases [3]. A prospective study by European researchers in 2022 reported that up to 33.3% of gene variants associated with Noonan syndrome were found in fetuses with NIHF and a normal karyotype [2]. These findings likely reflect a more severe phenotypic manifestation of the disease at the antenatal stage in the study participants.
Some of the primary manifestations of Noonan syndrome during the antenatal stage include polyhydramnios, increased fetal nuchal translucency thickness, neck cystic hygroma, generalized edema, hydrothorax, and NIHF [12]. A combination of these pathologies, with fetal malformations of the urinary system and congenital heart defects, such as VSD, ASD, or coarctation of the aorta, is possible. However, according to various sources, 68–94% of heart defects are diagnosed later in childhood [12]. In our study, only one case exhibited additional characteristic findings (hypertelorism, pyelectasis, and VSD), whereas the remaining cases presented with isolated NIHF. These data underscore the clinical significance of whole-exome sequencing in all cases of prenatally diagnosed NIHF [13].
A retrospective study conducted by a group of Chinese scientists in 2021 described the clinical manifestations of Noonan syndrome during the antenatal period, similar to our observation of a diagnosed variant in the LZTR1 gene, including increased fetal nuchal translucency thickness from 13 to 14 weeks [12]. The Noonan syndrome phenotype described by Miceikaite I. et al. (2021) in fetuses with an established de novo RIT1 variant mirrors the clinical findings of clinical case 1. During the first-trimester prenatal screening, neck cystic hygroma was identified as the only manifestation of NIHF. Bilateral pyelectasis and hydrothorax were subsequently diagnosed at 18 and 28 weeks, respectively [14].
In our country, extended genetic testing is not performed in all cases during the antenatal stage, or in newborns with developmental defects. Diagnosis depends on the severity of the clinical manifestations, but it can be challenging owing to the wide variability of symptoms. Uncomplicated cases may go undiagnosed and can only be identified in adulthood or after the birth of a child with more severe clinical manifestations in the family.
When diagnosing variants associated with Noonan syndrome in the fetus, it is necessary to examine parents and other family members to assess the pathogenicity and inheritance pattern of the variant(s) [3, 4]. In cases where a de novo variant associated with autosomal dominant inheritance is diagnosed in the fetus, the risk of recurrence of Noonan syndrome is less than 5%. This information is reassuring for parents as it indicates that the adverse pregnancy outcome was due to a random genetic event. Couples with a de novo inheritance variant should be counseled for future pregnancy planning.
In clinical case 3 of our study, Noonan syndrome with autosomal recessive inheritance, caused by variants in the LZTR1 gene inherited from both parents, was identified. During genetic counseling, it is essential to inform the family of the 25% risk of disease recurrence in future pregnancies. This information broadens the couple's reproductive choices, including the option of in vitro fertilization with preimplantation genetic testing for monogenic diseases, allowing for the selection of a viable embryo in future pregnancies. In cases of spontaneous pregnancy, early invasive prenatal diagnosis and targeted genetic testing for familial variants using Sanger sequencing can be performed at 9–10 weeks of gestation to rule out homozygous or compound heterozygous variant carriage in the fetus.
Early and accurate diagnosis of Noonan syndrome is crucial, as it affects pregnancy management and the prognosis of the fetus's life and health [15]. Prenatal diagnosis can be performed using chorionic villus sampling or amniocentesis. Early diagnosis enables parents to be informed about the possible disease course, life expectancy, treatment options, and potential long-term outcomes for their child. In severe cases of Noonan syndrome, the diagnosis can be made antenatally as early as 9 weeks of gestation. This information is vital for families to decide whether to continue or terminate the pregnancy [4, 16, 17]. Moreover, testing family members can help establish the pathogenicity of the variant and guide the prognosis of future pregnancies in the family.
Although in most patients with autosomal dominant Noonan syndrome, the variant occurs de novo, in 20–40% of cases, it is inherited from one of the parents, more commonly the mother. In most cases, parents with mild forms of Noonan syndrome are unaware of their genetic status. Detecting inherited variants helps clarify the risk of Noonan syndrome recurrence in future offspring and enables informed reproductive decisions and pregnancy planning [2, 18].
Conclusion
In summary, whole-exome sequencing may aid in diagnosing Noonan syndrome in cases of NIHF, allowing for more targeted counseling regarding both the likely pregnancy outcome and the long-term health and developmental prospects for the child. Testing family members may help to assess prognosis, counsel the family on the potential presentation of the disease, and guide the couple's reproductive choices when planning a subsequent pregnancy. Advanced genetic testing may facilitate early assessment of the fetus in future pregnancies before prenatal screening and the onset of NIHF symptoms, or enable referral to a fertility specialist for preimplantation genetic testing for a single-gene disorder to help ensure a healthy child.
References
- Roberts A.E., Allanson J.E., Tartaglia M., Gelb B.D. Noonan syndrome. Lancet. 2013; 381(9863): 333-42. https://dx.doi.org/10.1016/S0140-6736(12)61023-X.
- Zenker M., Edouard T., Blair J.C., Cappa M. Noonan syndrome: improving recognition and diagnosis. Arch. Dis. Child. 2022; 107(12): 1073-8. https://dx.doi.org/10.1136/archdischild-2021-322858.
- Sparks T.N., Lianoglou B.R., Adami R.R., Pluym I.D., Holliman K., Duffy J. et al.; University of California Fetal-Maternal Consortium; University of California, San Francisco Center for Maternal-Fetal Precision Medicine. Exome sequencing for prenatal diagnosis in nonimmune hydrops fetalis. N. Engl. J. Med. 2020; 383(18): 1746-56. https://dx.doi.org/10.1056/NEJMoa2023643.
- Carcavilla A., Suárez-Ortega L., Rodríguez Sánchez A., Gonzalez-Casado I., Ramón-Krauel M., Labarta J.I. et al. Síndrome de Noonan: actualización genética, clínica y de opciones terapéuticas [Noonan syndrome: genetic and clinical update and treatment options]. An. Pediatr. (Engl. Ed.). 2020; 93(1): 61.e1-61.e14. (in Spanish). https://dx.doi.org/10.1016/j.anpedi.2020.04.008.
- Tajan M., Paccoud R., Branka S., Edouard T., Yart A. The RASopathy family: consequences of germline activation of the RAS/MAPK pathway. Endocr. Rev. 2018; 39(5): 676-700. https://dx.doi.org/10.1210/er.2017-00232.
- Grant A.R., Cushman B.J., Cavé H., Dillon M.W., Gelb B.D., Gripp K.W. et al. Assessing the gene-disease association of 19 genes with the RASopathies using the ClinGen gene curation framework. Hum. Mutat. 2018; 39(11): 1485-93. https://dx.doi.org/10.1002/humu.23624.
- Capri Y., Flex E., Krumbach O.H.F., Carpentieri G., Cecchetti S., Lißewski C. et al. Activating mutations of RRAS2 are a rare cause of Noonan syndrome. Am. J. Hum. Genet. 2019; 104(6): 1223-32. https://dx.doi.org/10.1016/j.ajhg.2019.04.013.
- Myers A., Bernstein J.A., Brennan M.L., Curry C., Esplin E.D., Fisher J. et al. Perinatal features of the RASopathies: Noonan syndrome, cardiofaciocutaneous syndrome and Costello syndrome. Am. J. Med. Genet. A. 2014; 164A(11):2814-21. https://dx.doi.org/10.1002/ajmg.a.36737.
- Stuurman K.E., Joosten M., van der Burgt I., Elting M., Yntema H.G., Meijers-Heijboer H. et al. Prenatal ultrasound findings of rasopathies in a cohort of 424 fetuses: update on genetic testing in the NGS era. J. Med. Genet. 2019; 56(10): 654-61. https://dx.doi.org/10.1136/jmedgenet-2018-105746.
- Al-Kouatly H.B., Shivashankar K., Mossayebi M.H., Makhamreh M., Critchlow E., Gao Z. et al. Diagnostic yield from prenatal exome sequencing for non-immune hydrops fetalis: a systematic review and meta-analysis. Clin. Genet. 2023; 103(5): 503-12. https://dx.doi.org/10.1111/cge.14309.
- Гандаева Л.А., Каверина В.Г., Басаргина Е.Н., Пушков А.А., Савостьянов К.В. Редкий случай синдрома Нунан, обусловленный биаллельными вариантами в гене LZTR1. Неврологический журнал имени Л.О. Бадаляна. 2023; 4(3): 120-9. [Gandaeva L.A., Kaverina V.G., Basargina E.N., Pushkov A.A., Savost’yanov K.V. A rare case of Noonan syndrome associated with biallelic variants in the LZTR1. Neurological Journal named after L.O. Badalyan. 2023; 4(3): 120-9. (in Russian)]. https://dx.doi.org/10.46563/2686-8997-2023-4-3-120-129.
- Menashe M., Arbel R., Raveh D., Achiron R., Yagel S. Poor prenatal detection rate of cardiac anomalies in Noonan syndrome. Ultrasound Obstet. Gynecol. 2002; 19(1): 51-5. https://dx.doi.org/10.1046/j.0960-7692.2001.00485.x.
- Emms A., Castleman J., Allen S., Williams D., Kinning E., Kilby M. Next generation sequencing after invasive prenatal testing in fetuses with congenital malformations: prenatal or neonatal investigation. Genes (Basel). 2022; 13(9): 1517. https://dx.doi.org/10.3390/genes13091517.
- Tano S., Kotani T., Yoshihara M., Nakamura N., Matsuo S., Ushida T. et al. A case of non-immune hydrops fetalis with maternal mirror syndrome diagnosed by trio-based exome sequencing: an autopsy case report and literature review. Mol. Genet. Metab. Rep. 2022; 33: 100925. https://dx.doi.org/10.1016/j.ymgmr.2022.100925.
- Bhambhani V., Muenke M. Noonan syndrome. Am. Fam. Physician. 2014; 89(1): 37-43.
- Quinn A.M., Valcarcel B.N., Makhamreh M.M., Al-Kouatly H.B., Berger S.I. A systematic review of monogenic etiologies of nonimmune hydrops fetalis. Genet. Med. 2021; 23(1): 3-12. https://dx.doi.org/10.1038/s41436-020-00967-0.
- Глотов О.С., Чернов А.Н., Глотов А.С., Баранов В.С. Перспективы применения экзомного секвенирования для решения проблем в репродукции человека (часть II). Акушерство и гинекология. 2022; 12: 40-5. [Glotov O.S., Chernov A.N., Glotov A.S., Baranov V.S. Prospects for using exome sequencing to solve problems in human reproduction (Part II). Obstetrics and Gynecology. 2022; (12): 40-5 (in Russian)]. https://dx.doi.org/10.18565/aig.2022.220.
- Глотов О.С., Чернов А.Н., Глотов А.С., Баранов В.С. Перспективы применения экзомного секвенирования для решения проблем в репродукции человека (часть I). Акушерство и гинекология. 2022; 12: 34-9. [Glotov O.S., Chernov A.N., Glotov A.S., Baranov V.S. Prospects for using exome sequencing to solve problems in human reproduction (Part I). Obstetrics and Gynecology. 2022; (12): 34-9. (in Russian)]. https://dx.doi.org/10.18565/aig.2022.221.
Received 06.08.2024
Accepted 27.10.2024
About the Authors
Daria G. Lyushnina, PhD student, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, +7(906)308-60-78,d_lyushnina@oparina4.ru, https://orcid.org/0009-0004-3160-8737
Jekaterina Shubina, PhD in Biology, Head of the Laboratory of Genomic Data Analysis, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia,
4, Acad. Oparin str., Moscow, Russia, 117997, +7(495)531-44-44, e_shubina@oparina4.ru, https://orcid.org/0000-0003-4383-7428
Nana K. Tetruashvili, PhD, Head of the Obstetric Department of Pregnancy Pathology No. 2, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia,
4, Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-14-77, n_tetruashvili@oparina4.ru, https://orcid.org/0000-0002-9201-2281
Nadezhda V. Zaretskaya, PhD, Head of the Laboratory of Clinical Genetics of the Department of Clinical Genetics, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-24-11, znadezda@yandex.ru, https://orcid.org/0000-0001-6754-3833
Anna S. Bolshakova, MD, Geneticist at the Department of Clinical Genetics, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-24-11, a_bolshakova@oparina4.ru, https://orcid.org/0000-0002-7508-0899
Viktoriia S. Pak, PhD student, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, +7(913)897-28-49,
v_pak@oparina4.ru, https://orcid.org/0009-0002-1444-9071
Maria V. Kuznetsova, PhD in Biology, Senior Researcher at the Laboratory of Molecular and Genetic Methods of the Institute of Reproductive Genetics, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-13-41, mkarja@mail.ru, https://orcid.org/0000-0003-3790-0427
Galina V. Mikhailovskaya, Biologist at the Laboratory of Molecular and Genetic Methods of the Institute of Reproductive Genetics, V.I. Kulakov NMRC for OG&P,
Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-13-41, g_mikhailovskaia@oparina4.ru
Ekaterina L. Bokeriya, PhD, Researcher at the Department of Pathology for Newborn and Prematurely-Born Children No. 2, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-27-05, e_bokeriya@oparina4.ru, https://orcid.org/0000-0002-8898-9612
Dmitrii Yu. Trofimov, PhD in Biology, Head of the Department of Clinical Genetics, V.I. Kulakov NMRC for OG&P, Ministry of Health of Russia, 4, Acad. Oparin str., Moscow, Russia, 117997, +7(495)438-49-51, d_trofimov@oparina4.ru, https://orcid.org/0000-0002-1569-8486
Corresponding author: Daria G. Lyushnina, d_lyushnina@oparina4.ru
Similar Articles

