

Exome sequencing for diagnosing and predicting uterine and vaginal anomalies
Objective: To identify genetic markers of uterine and vaginal malformations, including complex and rare combinations of other organs and systems abnormalities, by analyzing gene expression and methylation.Arakelyan A.S., Shubina Je., Popryadukhin A.Yu., Goltsov A.Yu., Trofimov D.Yu., Adamyan L.V.
Materials and methods: Exome sequencing was performed in 21 patients who underwent surgery for uterine and vaginal malformations at the V.I. Kulakov NMRC for OG&P from 2016 to 2020.
Results: The study identified pathogenic, probably pathogenic variants, and variants of the unclear significance of the NPHP3, TMEM67, ROBO2, MYRF, TRP6, SRCAP, PROKR2, SPRY4, and GREB1L genes with a high degree of probability associated with the pathogenesis of uterine and vaginal malformations.
Conclusion: The study findings suggest the role of genetic factors in the pathogenesis of uterine and vaginal malformations. Allelic variants of the discovered candidate genes associated with uterine and vaginal malformations can be used as markers to assess the risks of developing reproductive system anomalies. Further studies are required to identify the causes of uterine and vaginal malformations. Genetic studies of the patient's parents may provide additional information on the etiology of the disease.
Keywords
aplasia of the uterus and vagina
genes
exome
exome sequencing
malformations of the uterus and vagina
Uterine and vaginal anomalies are congenital malformations characterized by morphological changes occurring during a critical developmental period of the reproductive tract and resulting in impaired female reproductive function, including infertility, dysmenorrhea, and miscarriage. Currently, the incidence of malformations of uterine and vaginal anomalies ranges from 4 to 7% [1–4].
Although genital abnormalities may be isolated, there is a large number of combinations of congenital malformations of the female genital tract and other organ abnormalities, including kidney defects (25%), skeletal anomalies (10–12%), and, in rare cases, anomalies of the heart and sensory organs [5–7].
Congenital malformations of the female genital tract are associated with impaired fertility, miscarriages, and other adverse obstetric outcomes, as well as primary amenorrhea, pain syndrome, and prevent sexual activity. Considering this, it is essential to understand the pathogenesis and risk factors for the development of female genital abnormalities and their impact on women's reproductive health [8, 9]. Current understanding, based on the latest evidence, allows for thorough, accurate, and effective examination and treatment of patients with uterine and vaginal malformations.
Genital malformations are multifactorial. Many researchers believe that they can result from hereditary (endogenous) and environmental (exogenous) factors [10–13].
According to the literature, harmful exogenous factors, including hypoxia, hyper- and hypothermia, ionizing radiation, chemical compounds, pathogenic microbes, and alcohol, can adversely affect the embryo and fetus [14, 15].
Familial occurrence of uterine and vaginal aplasia described in the literature may indicate a hereditary etiology of this disease. However, an accurate assessment of familial forms is difficult due to incomplete diagnostic examination, lack of analysis of the family tree, and appropriate screening of siblings of patients [16–18]. Another factor may be the lack of clinical information about family members (pelvic ultrasound and magnetic resonance imaging, asymptomatic disease in women with minor anomalies that do not reduce the quality of life), which may be due to the lack of documented incidence of familial uterine and vaginal aplasia.
The data available today indicate a significant role of genetic factors in developing the disease [19–21]. According to various authors, 13–25% of congenital malformations are caused by genetic factors, multifactorial disorders caused by the sum of genetic and environmental factors, and chromosomal mutations such as translocations, deletions, duplications, and inversions. Ten percent of congenital disorders are caused by external factors, and in 65% of cases, the cause of the diseases cannot be established [22].
Despite extensive research in reproductive genetics and advancements in genome analysis, genetic heterogeneity in uterine and vaginal malformations is probably underestimated because most of the research to date is based on candidate genes. Broader techniques such as complete exome sequencing, especially in familial cases, should be applied to identify new causative genes and clarify the pattern of inheritance and penetrance. Besides, discovering new genes that cause the Mayer–Rokitansky–Küster-Hauser syndrome (MRKH) will also increase knowledge about the molecular mechanisms underlying female reproductive tract development. Only the analysis of large cohorts of patients with uterine and vaginal anomalies, in particular MRKH, will help identify new candidate genes and establish the phenotype/genotypic correlations necessary for genetic analysis, diagnosis of uterine and vaginal abnormalities, as well as identifying risk factors for the development of these anomalies.
The present study aimed to identify genetic markers of uterine and vaginal malformations, including complex and rare combinations of other organs and systems abnormalities, by analyzing gene expression and methylation.
The study was conducted as per the Helsinki Declaration and principles of Good Clinical Practice. The study protocol was approved by the Ethics Committees of all participating clinical centers. Written informed consent was obtained from all participants before enrollment.
Materials and methods
We conducted a genetic analysis of 15 women with uterine and vaginal aplasia, three with bladder exstrophy, one with partial vaginal aplasia with a functioning unicornuate uterus, and two with and double uterus and vagina with partial aplasia of the second vagina accompanied by other severe anomalies, such as the impaired formation of the musculoskeletal system or its parts, malformations of the urinary system (renal aplasia, urinary bladder exstrophy), hearing impairment, etc.
The patients underwent surgical correction of malformations at the Department of Operative Gynecology; the laboratory investigations were conducted at the Institute of Reproductive Genetics of the V.I. Kulakov NMRC for OG&P of Minzdrav of Russia. Blood samples and phenotypic data were collected. Written informed consent was obtained from all subjects. All patients had a balanced karyotype 46, XX. Peripheral blood samples were taken from each patient. DNA from blood cells was isolated using a QIAamp DNA Blood Mini Kit (QIAGEN). Sequencing library preparation using target enrichments was performed using SureSelectXT V7 enrichment kits according to the manufacturer's instructions.
Sequencing was performed on an MGISeq-2000 sequencer according to the manufacturer's instructions. Alignment to the reference genome (version hg38) and the search for differences from the reference genome was performed with the GATK software using the GATK best practices [23]. The obtained variants were annotated using the Variant effect predictor tool; the annotated variants were filtered using scripts developed by the authors [24]. The gene analysis associated with the female genital tract malformations and the genitourinary system was carried out. We used the list of genes from the Human phenotype ontology database to analyze the data: https://hpo.jax.org/app/.
Results
The study findings demonstrated a possible relationship with the phenotype in 9/21 (42%) cases. The results of the analysis of variants are shown in the table. In 7 samples, probably pathogenic variants and variants of unclear clinical significance were found in genes associated with dominant diseases (ROBO2, MYRF, TRPC6, SRCAP, GREB1L, SPRY4, PROKR2). A genetic examination of the patient's parents is required to determine the clinical significance.
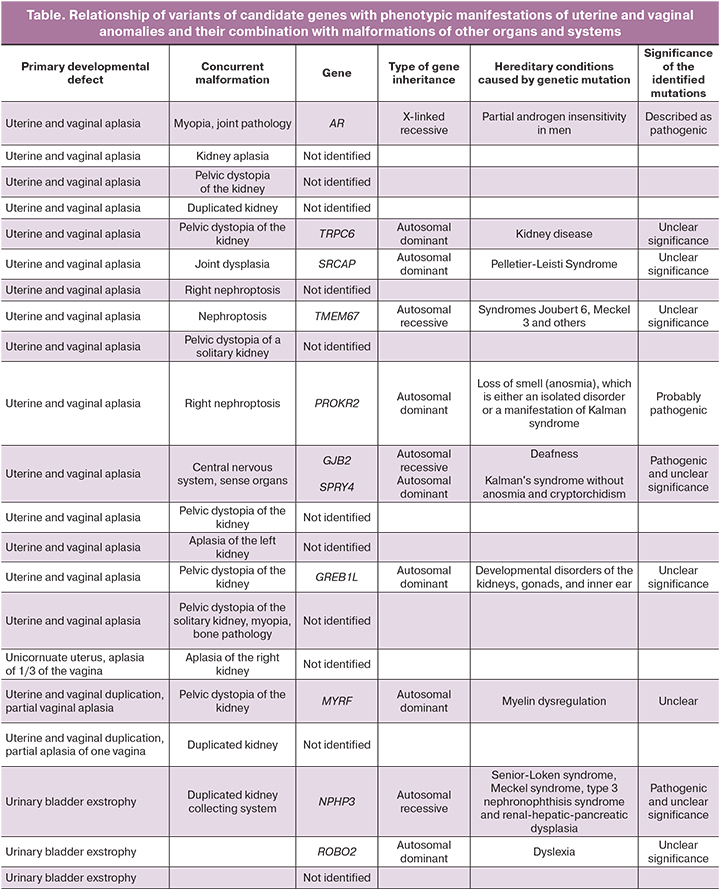
In two cases, variants associated with autosomal recessive syndromes (NPHP3 and TMEM67) were found; their clinical presentations included nephroptosis, impaired kidney and liver development.
Segregation analysis is required to clarify the significance. If the variants are inherited from both parents, and their significance is established, there is a 25% chance of having a second child with the same disorder in the family.
In one case, a pathogenic homozygous variant was additionally found in the GJB2 gene associated with deafness, which was consistent with the features observed in the patient but did not explain uterine aplasia.
In one case, a previously described pathogenic variant was found in the gene for the androgen receptor AR in a heterozygous state, leading to partial androgen insensitivity in men.
Discussion
The cause of uterine and vaginal anomalies remains unknown. Many researchers consider Müllerian duct anomalies of polygenic or multifactorial origin involving complex genetic mechanisms [4, 8, 25, 26].
Of most significant scientific interest is MRKH, also referred to as uterine and vaginal aplasia, the second most common cause of primary amenorrhea and is associated with sexual activity problems. It is characterized by the congenital agenesis of the uterus, cervix, and vagina in phenotypically normal girls with a 46, XX karyotype.
Agenesis of female genital organs can occur in combination with other rare syndromes, such as McKusick–Kaufman syndrome (MKKS gene, locus 20p12), Bardet – Biedl syndrome (MKKS gene, locus 20p12, and some other genes in different chromosomes), Wolf–Hirschhom syndrome (deletions of chromosome 4p16.3), Goldenhar syndrome, which may indicate a common origin of etiological factors [27].
The heterogeneity of MRKH suggests the presence of molecular defects in internal organs development that are closely related during embryogenesis. Indeed, MRKH manifests itself due to damage at 5–6 weeks of gestation, affecting the intermediate mesoderm and leading to fusion of the Müllerian ducts. The renal system also develops from the mesoderm, which explains the renal agenesis or ectopia often associated with MRKH [28].
For a long time, the syndrome was considered a sporadic anomaly, but the increasing number of familial aggregates supports the hypothesis of a genetic cause [17, 29]. In familial cases, the syndrome may be transmitted as autosomal dominant with incomplete penetrance and variable expressivity. This suggests the involvement of either mutation in the underlying developmental gene or a more limited chromosomal imbalance. According to the author, only 68 cases of familial MRKH were registered [17].
According to L.V. Adamyan et al. (2008), genetic factors play an essential role in the embryogenesis of uterine and vaginal malformations and the etiopathogenetic mechanisms of the formation of endometriotic lesions [19]. Analyzing information and various clinical cases of the combination of MRKH with external and internal endometriosis, the group of authors identified the relationship between these diseases and suggested that they are multifactorial diseases, the causes of which may be genetic polymorphisms, heredity, hormonal effects on estrogen and progesterone receptors [30].
The critical role of WNT, HOXA, and PAX genes playing an essential role in embryogenesis is widely discussed in the literature [31–33].
The WNT family includes a group of genes involved in embryonic development. Moreover, the WNT genes play a notable role in developing the mammalian genitourinary system [34, 35].
The homeobox genes belong to a large family of HOX clusters. Some of the HOX genes (HOXA9-13 and HOXB9-13) play a crucial role in developing the female reproductive tract and are therefore considered putative MRKH genes [36].
Surprisingly, deletion of the entire HOXA cluster does not cause more urogenital anomalies than single monoallelic HOXA13 mutations [37].
HOXA9 is expressed in the fallopian tubes, HOXA10 in the uterus, HOXA11 in the uterus and cervix, and HOXA13 in the upper part of the vagina [33, 38].
Genes with a broad spectrum of activity in early development (e.g., WT1 [38], PAX2, HOXA7 in HOXA13, and PBX1 [39]) have also been proposed as candidates based on the observed phenotypes in mutant mice [33]. However, their role in MRKH has not subsequently been demonstrated.
Of scientific interest are the data that in monozygotic twins, one develops MRKHS and the other does not, which is due to differences in phenotype. This indicates that the pathogenesis of MRKHS is associated with epigenetic mechanisms linked to environmental and stochastic factors [18]. Using genome-wide analysis, Rall et al. [40] investigated the differences in transcription products and methylation levels between patients with MRKH and healthy volunteers. By evaluating two gene clusters, nine potentially causative genes (HOXA5, HOXA9, WISP2, CDH5, PEG10, MFAP5, LRRC32, RALGPS2, and ralgps2) were identified. Six of these genes (CDH5, MFAP5, WISP2, HOXA5, PEG10, and HOXA9) are involved in developing female genitalia. Subsequent network analyses identified WISP2, HOXA5, HOXA9, GATA4, and WT1 as critical genes in MRKHS.
In their study, De Tomasi L. et al. described cases of females with uterine and renal aplasia, confirming that the GREB1L gene plays a vital role in developing the kidneys and female reproductive tract [41]. Morten K. Herlin et al. state that GREB1L is a new and promising candidate gene in the etiology of MRKH [42].
Interestingly, genital malformations such as a bicornuate uterus [43] and the Müllerian duct aplasia [44] were sometimes associated with renal abnormalities in some familial cases that demonstrated mutations in the TCF2 gene. Defects in this gene may thus explain some rare cases of genital malformations, including aplasia, making this gene one of the candidates for a genetic link to MRKH, but is limited to familial cases with renal and/or diabetic history.
Other authors reported that a significant part of sexual development disorders is etiologically associated with chromosomal abnormalities and affects chromosomes 1-7, 10-18, 22, and X. But a comparison of the results of different studies revealed only five repeated deletions/duplications in chromosomal regions 1q21.1, 16p11.2, 17q12, 22q11.21 and Xp22 [45–50]. Overall, these changes were found in 28 MRKH patients and accounted for approximately 10% of MRKH cases.
Phenotypic data indicate that associated renal, skeletal, and other congenital anomalies occur in our cohort. We have identified new areas of genomic imbalance in women with genital anomalies and other malformations of the Müllerian duct, further elucidating the complex genetic architecture of these conditions.
The growing number of familial cases and the nature of congenital malformations indicate that uterine and vaginal aplasia is a disorder that arises during embryonic development and genetic factors play a role in establishing the condition [27, 51]. We found in 7 cases variants in genes (ROBO2, MYRF, TRPC6, SRCAP, GREB1L, SPRY4, PROKR2) of probable pathogenic and unclear significance associated with dominant diseases. Mutations in the MYRF gene, which encodes a transcription factor necessary for the differentiation of oligodendrocytes and is related to the development of cardiovascular syndrome, can lead to the absence of the uterus and ovaries. GREB1L has been reported as a target of retinoic acid signaling, and the dysregulation of which results in renal aplasia/hypoplasia. Mutations in this gene can lead to malformations of the uterus, vagina, and ovaries; the phenotype can be highly variable, and penetrance is incomplete. Mutations of ROBO2 have been associated with impaired renal and urinary tract development. SRCAP encodes the ATPase required to insert histone H2A into the nucleosome. Mutations in this gene lead to the Floating–Harbor syndrome, manifesting as abnormalities in the development of the genitals and joints. The PROKR2 gene encodes an intramembrane protein and receptor for prokineticins. SPRY4 is an inhibitor of the receptor-transduced mitogen-activated protein kinase signaling pathway. Mutations in these genes are associated with hypogonadotropic hypogonadism, primary amenorrhea, and the absence of puberty. A mutation in the TRP6 gene is linked to impaired renal function and abnormal morphology; it does not lead to uterine and vaginal aplasia but can modify the clinical picture. In this situation, it is necessary to study variants more extensively to a variant in the genes. If the variants arose de novo and the significance of the variants is confirmed, then the chance of having a second child with the same disorder in the family is 1–2% (one cannot exclude gonadal mosaicism in the parents). However, these women can give birth to a genetically normal child with a 50% risk of passing on the mutation to the child.
The NPHP3 and TMEM67 genes are associated with autosomal recessive syndromes, including nephronophthisis, renal-hepatic-pancreatic dysplasia, and Meckel syndrome; clinical presentations include nephroptosis and impaired kidneys and liver development.
The previously described pathogenic variant in the gene for the androgenic receptor AR in the heterozygous state, which we discovered in one case, is an X-linked gene. Still, given that this variant in the gene can cause an androgen insensitivity syndrome in men, its role in the formation of uterine and vaginal anomalies is not excluded. Therefore, when this woman gives birth to a boy after conceiving using assisted reproductive technologies and surrogacy, the probability of having a child with androgen insensitivity syndrome is 50%. Thus, women with uterine and vaginal malformations seeking surrogacy should be counseled about preimplantation genetic diagnosis, given the potential risk of passing on such a genomic imbalance to their offspring.
The study of DNA exomes in patients with uterine and vaginal malformations can support the genetic hypothesis of developing a complex genital defect. Not all variants identified in this study were associated with the clinical presentation corresponding to gene mutations. To obtain accurate data and elucidate the complex underlying defects of this heterogeneous disease, it is necessary to analyze many patients and conduct a functional analysis of the disease.
Conclusion
The present study was conducted to identify common genetic causes of complex and rare malformations in patients with congenital malformations of the female genital organs. Determining the potential underlying causes of genital malformations requires a careful and detailed analysis of the history and many genes. Genomic analysis of gene expression and methylation can help understand molecular pathways. Identifying the cause of the developmental defects will enable a more accurate diagnosis and assessment of the risks of having a second child with the same developmental defects and conduct the necessary prenatal testing. Patients with uterine and vaginal aplasia may likely have heterozygous de novo mutations in yet-unknown genes. We believe that genetic methods can increase the chances of identifying the causative genes for this malformation. In our opinion, it is necessary to test all family members and all family cases to determine the patterns of inheritance and establish correlations between genotype and phenotype and assess the risks of having another child with the same developmental defects in the family.
References
- Grimbizis G.F., Gordts S., Di Spiezio S.A., Brucker S., De Angelis C., Gergolet M. et al. The ESHRE/ESGE consensus on the classification of female genital tract congenital anomalies. Hum Reprod. 2013; 28(8): 2032-44. https://dx,doi.org/10.1093/humrep/det098.
- Chan Y.Y., Jayaprakasan K., Zamora J., Thornton J.G., Raine-Fenning N., Coomarasamy A. The prevalence of congenital uterine anomalies in unselected and high-risk populations: a systematic review. Hum. Reprod. Update. 2011; 17(6): 761-71. https://dx,doi.org/10.1093/humupd/dmr028.
- Saravelos S.H., Cocksedge K.A., Li T.C. Prevalence and diagnosis of congenital uterine anomalies in women with reproductive failure: a critical appraisal. Hum. Reprod. Update. 2008; 14(5): 415-29. https://dx,doi.org/10.1093/humupd/dmn018.
- Сафронов О.В., Брюхина Е.В., Ищенко Л.С., Сафронова Л.Е., Мшак-Манукян Г.Н. Современные классификационные системы и методологические подходы в диагностике аномалий развития матки. Акушерство и гинекология. 2019; 3: 18-24. [Safronov O.V., Briukhina E.V., Ishchenko L.S., Safronova L.E., Mshak-Manukyan G.N. Current classification systems and methodological approaches in the diagnosis of uterine malformations. Obstetrics and Gynecology. 2019; 3: 18-24. (in Russian)]. https://dx.doi.org/10.18565/aig.2019.3.18-24.
- Ribeiro S.C., Tormena R.A., Peterson T., Gonzales M., Serrano P.G., Almeida J.A.M., Baracat E. Müllerian duct anomalies: review of current management. Sao Paulo Med. J. 2009; 127: 92-6. https://dx,doi.org/10.1590/S1516-31802009000200007.
- Mueller G.C., Hussain H.K., Smith Y.R., Quint E.H., Carlos R.C., Johnson T.D., DeLancey J.O. Müllerian duct anomalies: comparison of MRI diagnosis and clinical diagnosis. AJR Am. J. Roentgenol. 2007; 189(6): 1294- 302. https://dx,doi.org/10.2214/AJR.07.2494.
- Кириллова Е.А., Курбанова А.Г., Трепаков Е.А. Клинико-генетические исследования при пороках развития мочеполовой системы женщины. В кн.: XIV Международный генетический конгресс. Секционные заседания. М.; 1978. [Kirillova E.A., Kurbanova A.G., Trepakov E.A. Clinical and genetic studies of malformations of the genitourinary system of women. In the book: XIV International Genetic Congress. Breakout sessions. M.; 1978. 322p. (in Russian)].
- Донников А.Е. Этические вопросы, связанные с преконцепционным генетическим скринингом: исторический опыт и современные тенденции. Акушерство и гинекология. 2019; 11: 46-54. [Donnikov A.E. Ethical issues related to preconceptional genetic screening: historical experience and current trends. Obstetrics and gynecology. 2019; 11: 46-54. (in Russian)]. https://dx.doi.org/10.18565/aig.2019.11.46-54.
- Буяновская О.А., Хохлова С.В., Сенча А.Н. Настоящее и будущее молекулярно-генетического анализа в скрининге и профилактике злокачественных новообразований репродуктивных органов у женщин. Акушерство и гинекология. 2019; 11: 55-64. [Buyanovskaya O.A., Khokhlova S.V., Sencha A.N. Present and future of molecular genetic analysis in screening and prevention of malignant neoplasms of reproductive organs in women. Obstetrics and gynecology. 2019; 11: 55-64. (in Russian)]. https://dx.doi.org/10.18565/aig.2019.11.55-64.
- Кобозева Н.В., Гуркин Ю.А. Реконструктивные операции при аномалиях у девочек. Современные методы оперативного лечения в акушерстве и гинекологии. М.; 1983. [Kobozeva N.V., Gurkin Yu.A. Reconstructive operations for anomalies in girls. Modern methods of surgical treatment in obstetrics and gynecology. M.; 1983. (in Russian)].
- Трепаков Е.А., Пиганова Н.Л. Медико-генетические аспекты аномалий развития матки. Акушерство и гинекология. 1973; 10: 44-8. [Trepakov E.A., Piganova N.L. Medical and genetic aspects of uterine malformations. Obstetrics and Gynecology. 1973; 10: 44-8. (in Russian)].
- Elias S., Carson S.A., Simpson J.A. The handfoot-uterus syndrome: a rare autosomal dominant disorder. Fertil. Steril. 1978; 29: 239.
- Golan A., Langer R., Bucovsky I. Congenital anomalies of the Mullerian fusion defects. Fertil. Steril. 1989; 51(5): 747-55.
- Хащенко Е.П., Аллахвердиева Э.З., Аракелян А.С., Уварова Е.В., Чупрынин В.Д., Кулабухова Е.А., Лужина И.А., Учеваткина П.В., Мамедова Ф.Ш., Асатурова А.В. Особенности дифференциальной диагностики и тактики ведения пациенток с редкой формой порока развития мюллеровых протоков ACUM и маточного рудиментарного рога в раннем репродуктивном возрасте. Акушерство и гинекология. 2021; 6: 156-67. [Khashchenko E.P., Allakhverdieva E.Z., Arakelyan A.S., Uvarova E.V., Chuprynin V.D., Kulabukhova E.A., Luzhina I.A., Uchevatkina P.V., Mamedova F.Sh., Asaturova A.V. Differential diagnosis and management tactics in patients with a rare developmental Müllerian duct anomaly ACUM and a rudimentary uterine horn at early reproductive age. Obstetrics and Gynecology. 2021; 6: 156-67. (in Russian)]. https://dx.doi.org/10.18565/aig.2021.6.156-167.
- Скосырева А.М., Балика Ю.Д., Карташева В.Е. Влияние этиолового алкоголя на развитие эмбриона и плода в эксперименте. Акушерство и гинекология. 1981; 1: 38-40. [Skosyreva A.M., Balika Yu.D., Kartasheva V.E. The effect of ethyl alcohol on the development of the embryo and fetus in the experiment. Obstetrics and Gynecology. 1981; 1: 38-40. (in Russian)].
- Shokeir M.H. Aplasia of the Müllerian system: evidence for probable sex- limited autosomal dominant inheritance. Birth Defects Orig. Artic Ser. 1978; 14(6C): 147-65.
- Herlin M., Højland A.T., Petersen M.B. Familial occurrence of Mayer- Rokitansky-Küster-Hauser syndrome: a case report and review of the literature. Am. J. Med. Genet. A. 2014: 164A(9): 2276-86. https://dx.doi.org/10.1002/ajmg.a.36652.
- Gervasini C., Grati F.R., Lalatta F., Tabano S., Gentilin B., Colapietro P. et al. SHOX duplications found in some cases with type I Mayer-Rokitansky-Kuster-Hauser syndrome. Genet. Med. 2010; 12(10): 634-40. https://dx.doi.org/10.1097/GIM.0b013e3181ed6185.
- Адамян Л.В., Спицын В.А., Андреева Е.Н. Генетические аспекты гинекологических заболеваний. М.: Медицина; 1998. [Adamyan L.V., Spitsyn V.A., Andreeva E.N. Genetic aspects of gynecological diseases. M.: Medicine; 1998. (in Russian)].
- Ma J., Qin Y., Liu W., Duan H., Xia M., Chen Z.J. Analysis of PBX1 mutations in 192 Chinese women with Müllerian duct abnormalities. Fertil. Steril. 2011; 95(8): 2615-7. https://dx.doi.org/10.1016/j.fertnstert.2011.04.074.
- Аракелян А.С., Попрядухин А.Ю., Карапетян Э.А. Генетические факторы развития синдрома Майера–Рокитанского–Кюстера–Хаузера (аплазии матки и влагалища). Проблемы репродукции. 2020; 26(5): 43-50. [Arakelyan A.S., Popryadukhin A.Yu., Karapetyan E.A. Genetic factors in the development of the Mayer–Rokitansky–Küster–Hauser syndrome (aplasia of the uterus and vagina). Problemy Reproduktsii (Russian Journal of Human Reproduction). 2020; 26(5): 43-50. (in Russian)]. https://dx.doi.org/10.17116/repro20202605143.
- Maneschi M., Maneschi F., Fuga G. Reproductive impairment of woman with unicornuate uterus. Acta Obstet. Gynecol. Scand. 1988; 67(6): 557-60.
- Van der Auwera G.A., O'Connor B.D. Genomics in the cloud: using Docker, GATK, and WDL in Terra. O'Reilly Media; 2020.
- McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A. et al. The ensembl variant effect predictor. Genome Biol. 2016; 17(1):122. https://dx.doi.org/10.1186/s13059-016-0974-4.
- Cheroki C., Krepischi-Santos A.C., Szuhai K., Brenner V., Kim C.A., Otto P.A., Rosenberg C. Genomic imbalances associated with Müllerian aplasia. J. Med. Genet. 2008; 45(4): 228-32. https://dx.doi.org/10.1136/jmg.2007.051839.
- Schnabel C.A., Selleri L., Cleary M.L. Pbx1 is essential for adrenal development and urogenital differentiation. Genesis. 2003; 37: 123-30.
- Бобкова М.В., Баранова Е.Е., Кузнецова М.В., Трофимов Д.Ю., Адамян Л.В. Семейный случай синдрома Мейера-Рокитанского-Кюстера-Хаузера и обзор литературы. Проблемы репродукции. 2015; 21(4): 17-22. [Bobkova M.V., Baranova E.E., Kuznetsova M.V., Trofimov D.Yu., Adamyan L.V. Family case of Mayer-Rokitansky-Kuster-Hauser syndrome and literature review. Russian Journal of Human Reproduction. 2015; 21(4): 17-22. (in Russian)]. https://dx.doi.org/10.17116/repro201521417-22.
- Ludwig K.S. The Mayer-Rokitansky-Küster syndrome. An analysis of its morphology and embryology. Part II: Embryology. Arch. Gynecol. Obstet. 1998; 262(1-2): 27-42.
- Guerrier D., Mouchel T., Pasquier L., Pellerin I. The Mayer-Rokitansky-Küster-Hauser syndrome (congenital absence of uterus and vagina)-phenotypic manifestations and genetic approaches. J. Negat. Results Biomed. 2006; 5: 1. https://dx.doi.org/10.17116/repro201521417-22.10.1186/1477-5751-5-1.
- Адамян Л.В., Попрядухин А.Ю., Аракелян А.С., Козаченко И.Ф., Фархат К.Н. Аплазия матки и влагалища (синдром Майера-Рокитанского-Кюстера-Хаузера) в сочетании с эндометриозом: нерешенные аспекты этиологии и патогенеза (обзор литературы). Проблемы репродукции. 2016; 22(3): 8-14. [Adamyan L.V., Popryadukhin A.Yu., Arakelyan A.S., Kozachenko I.F., Farkhat K.N. Aplasia of the uterus and vagina (Mayer-Rokitansky-Kuster-Hauser syndrome) in combination with endometriosis: unresolved aspects of etiopathogenesis (a review). Russian Journal of Human Reproduction. 2016; 22(3):8-14. (in Russian)]. https://dx.doi.org/10.17116/repro20162238-14.
- Ma W., Li Y., Wang M., Li H., Su T., Li Y., Wang S. Associations of polymorphisms in WNT9B and PBX1 with Mayer-Rokitansky-Küster-Hauser syndrome in Chinese Han. PLoS One. 2015; 10(6): e0130202. https://dx.doi.org/10.1371/journal.pone.0130202.
- Kim J.J., Taylor H.S., Lu Z., Ladhani O., Hastings J.M., Jackson K.S., Fazleabas A.T. Altered expression of HOXA10 in endometriosis: potential role in decidualization. Mol. Hum. Reprod. 2007; 13(5): 323-32. https://dx.doi.org/10.1093/molehr/gam005.
- Бобкова М.В., Баранова Е.Е., Адамян Л.В. Генетические аспекты формирования аплазии влагалища и матки: история изучения. Проблемы репродукции. 2015; 21(3): 10-5. [Bobkova M.V., Baranova E.E., Adamyan L.V. Genetic aspects of vagina and the uterus aplasia: the history. Russian Journal of Human Reproduction. 2015; 21(3): 10-15. (in Russian)]. https://dx.doi.org/10.17116/repro201521310-15.
- Carroll T.J., Park J.S., Hayashi S., Majumdar A., McMahon A.P. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell. 2005; 9(2): 283-92. https://dx.doi.org/10.1016/j.devcel.2005.05.016.
- Halt K., Vainio S. Coordination of kidney organogenesis by Wnt signaling. Pediatr. Nephrol. 2014; 29(4): 737-44. https://dx.doi.org/10.1007/s00467-013-2733-z.
- Massé J., Watrin T., Laurent A., Deschamps S., Guerrier D., Pellerin I. The developing female genital tract: from genetics to epigenetics. Int. J. Dev. Biol. 2009: 53(2-3): 411-24. https://dx.doi.org/10.1387/ijdb.082680jm.
- Devriendt K., Jaeken J., Matthijs G., Van Esch H., Debeer P., Gewillig M., Fryns J.P. Haploinsufficiency of the HOXA gene cluster, in a patient with hand-foot-genital syndrome, velopharyngeal insufficiency, and persistent patent Ductus botalli. Am. J. Hum. Genet. 1999; 65(1): 249-51. https://dx.doi.org/10.1086/302452.
- Taylor H.S., Vanden Heuvel G.B., Igarashi P. A conserved Hox axis in the mouse and human female reproductive system: late establishment and persistent adult expression of the Hoxa cluster genes. Biol. Reprod. 1997; 57(6):1338-45. https://dx.doi.org/10.1095/biolreprod57.6.1338.
- Burel A., Mouchel T., Odent S., Tiker F., Knebelmann B., Pellerin I., Guerrier D. Role of HOXA7 to HOXA13 and PBX1 genes in various forms of MRKH syndrome (congenital absence of uterus and vagina). J. Negat. Results Biomed. 2006; 5: 4. https://dx.doi.org/10.1186/1477-5751-5-4.
- Rall K., Barresi G., Walter M., Poths S., Haebig K., Schaeferhoff K. et al. A combination of transcriptome and methylation analyses reveals embryologically relevant candidate genes in MRKH patients. Orphanet. J. Rare Dis. 2011; 6: 32. https://dx.doi.org/10.1186/1750-1172-6-32.
- De Tomasi L., David P., Humbert C., Silbermann F., Arrondel C., Tores F. et al. Mutations in GREB1L cause bilateral kidney agenesis in humans and mice. Am. J. Hum. Genet. 2017; 101(5): 803-14. https://dx.doi.org/10.1016/j.ajhg.2017.09.026.
- Herlin M.K., Le V.Q., Højland A.T., Ernst A., Okkels H., Petersen A.C. et al. Whole-exome sequencing identifies a GREB1L variant in a three-generation family with Müllerian and renal agenesis: a novel candidate gene in Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome. A case report. Hum. Reprod. 2019; 34(9): 1838-46. https://dx.doi.org/10.1093/humrep/dez126.
- Bingham C., Ellard S., Cole T.R., Jones K.E., Allen L.I., Goodship J.A. et al. Solitary functioning kidney and diverse genital tract malformations associated with hepatocyte nuclear factor- 1beta mutations. Kidney Int. 2002; 61(4): 1243-51. https://dx.doi.org/10.1046/j.1523-1755.2002.00272.x.
- Lindner T.H., Njolstad P.R., Horikawa Y., Bostad L., Bell G.I., Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum. Mol. Genet. 1999; 8(11): 2001-8. https://dx.doi.org/10.1093/hmg/8.11.2001.
- Sundaram U.T., McDonald-McGinn D.M., Huff D., Emanuel B.S., Zackai E.H., Driscoll D.A., Bodurtha J. Primary amenorrhea and absent uterus in the 22q11.2 deletion syndrome. Am. J. Med. Genet. A. 2007; 43A(17): 2016-8. https://dx.doi.org/10.1002/ajmg.a.31736.
- Bernardini L., Gimelli S., Gervasini C., Carella M.., Baban A, Frontino G. et al. Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: two case reports. Orphanet J. Rare Dis. 2009; 4: 25. https://dx.doi.org/10.1186/1750-1172-4-25.
- Ledig S., Schippert C., Strick R., Beckmann M.W., Oppelt P.G., Wieacker P. Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-Küster-Hauser syndrome. Fertil. Steril. 2011: 95(5):1589-94. https://dx.doi.org/10.1016/j.fertnstert.2010.07.1062.
- Nik-Zainal S., Strick R., Storer M., Huang N., Rad R., Willatt L. et al. High incidence of recurrent copy number variants in patientswith isolated and syndromic Müllerian aplasia. J. Med. Genet. 2011: 48(3): 197-204. https://dx.doi.org/10.1136/jmg.2010.082412.
- Hinkes B., Hilgers K.F., Bolz H.J., Goppelt-Struebe M., Amann K., Nagl S. et al. A complex microdeletion 17q12 phenotype in a patient withrecurrent de novo membranous nephropathy. BMC Nephrol. 2012; 13: 27. https://dx.doi.org/10.1186/1471-2369-13-27.
- McGowan R., Tydeman G., Shapiro D., Craig T., Morrison N., Logan S. et al. DNA copy number variations are important in the complex genetic architecture of müllerian disorders. Fertil. Steril. 2015: 103(4): 1021-30.e1. https://dx.doi.org/10.1016/j.fertnstert.2015.01.008.
- Simpson J.L. Genetics of the female reproductive ducts. Am. J. Med. Genet. 1999; 89: 224-39.
Received 10.11.2021
Accepted 29.11.2021
About the Authors
Alek S. Arakelyan, PhD, Senior Researcher at the Gynecological Department, V.I. Kulakov National Medical Research Center of Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(916)376-78-98, arakelyanac@mail.ru, https://orcid.org/0000-0002-3217-1141, 117997, Russia, Moscow, Ac. Oparina str., 4.Jekaterina Shubina, Head of the Laboratory for Analysis of Genomic Data, V.I. Kulakov National Medical Research Center of Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)531-44-44, e_shubina@oparina4.ru, 117997, Russia, Moscow, Ac. Oparina str., 4.
Andrey Yu. Popryadukhin, PhD Student, V.I. Kulakov National Medical Research Center of Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, inferno_989@mail.ru, https://orcid.org/0000-0003-1466-8266, 117997, Russia, Moscow, Ac. Oparina str., 4.
Andrey Yu. Goltsov, Researcher at the Laboratory of Molecular Genetic Methods, V.I. Kulakov National Medical Research Center of Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)531-44-44, andrey.goltsov@gmail.com, 117997, Russia, Moscow, Ac. Oparina str., 4.
Dmitry Yu. Trofimov, Dr. Bio. Sci., Director of the Institute of Reproductive Genetics, V.I. Kulakov National Medical Research Center of Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, +7(495)438-49-51, d_trofimov@oparina4.ru, 117997, Russia, Moscow, Ac. Oparina str., 4.
Leila V. Adamyan, Academician of RAS, Dr. Med. Sci., Professor, Merited Scholar of the Russian Federation, Head Specialist in Gynecology of Minzdrav of Russia,
Head of the Department of Surgical Gynecology, V.I. Kulakov National Medical Research Center of Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia;
Head of the Department of Reproductive Medicine and Surgery, Faculty of Postgraduate Education, Moscow State University of Medicine and Dentistry,
+7(495)438-40-68, adamyanleila@gmail.com, https://orcid.org/0000-0002-3253-4512, 19997, Russia, Moscow, Ac. Oparina str., 4.
Corresponding author: Alek S. Arakelyan, arakelyanac@mail.ru
Authors' contributions: Arakelyan A.S., Popryadukhin A.Yu. – data collection and analysis; Trofimov D.Yu., Shubina Je., Goltsov A.Yu. – organization and conduct of laboratory investigations; Adamyan L.V., Arakelyan A.S., Popryadukhin A.Yu. – surgical treatment of patients; Arakelyan A.S., Trofimov D.Yu., Shubina Je. – drafting of the manuscript; Adamyan L.V., Arakelyan A.S., Trofimov D.Yu. – manuscript editing.
Conflicts of interest: The authors have no conflicts of interest to declare.
Funding: There was no funding for this study.
Patient Consent for Publication: All patients provided informed consent for the publication of their data.
Authors' Data Sharing Statement: The data supporting the findings of this study are available on request from the corresponding author after approval from the principal investigator.
For citation: Arakelyan A.S., Shubina Je., Popryadukhin A.Yu., Goltsov A.Yu., Trofimov D.Yu., Adamyan L.V. Exome sequencing for diagnosing and predicting uterine
and vaginal anomalies.
Akusherstvo i Ginekologiya/Obstetrics and Gynecology. 2021; 12: 120-127 (in Russian)
https://dx.doi.org/10.18565/aig.2021.12.120-127
Similar Articles

