

Genome-wide studies of uterine leiomyomas
Objective: This review aimed to investigate published genome-wide studies of uterine leiomyomas (ULs) and identify significant polymorphism loci linked to this condition using genome-wide association studies (GWAS).Alali O.M., Churnosov M.I.
Materials and methods: Relevant publications were searched in electronic databases such as PubMed, PubMed Central, and E-library, as well as in the GWAS catalog from 2011 to the present, using keywords such as uterine leiomyomas, fibroids, GWAS studies, and candidate genes.
Results: The results revealed eight genome-wide analyses of ULs, which identified 34 GWAS-significant polymorphic loci associated with disease generation and progression. However, findings from GWAS replication studies are limited and ambiguous. Among the known GWAS-significant genes, it is important to note that 15 out of the 34 genes were associated with the disease in only one GWAS, emphasizing the need for further (replicative) studies to explore the role of these genes in the development of the disease. Additionally, exon sequencing has provided valuable insights into the involvement of certain genes, such as MED12, in the formation of uterine fibroids, thereby expanding our understanding of the genetic determinants of the disease.
Conclusion: This review highlights the major genome-wide studies of ULs, identifies GWAS-significant polymorphisms associated with the disease, and emphasizes the potential utilization of these data in future replicative research and the enhancement of our knowledge about the molecular genetic mechanisms underlying the development of uterine fibroids.
Authors' contributions: Alali O.M., Churnosov M.I. – conception and design of the study; Alali O.M. – data collection and analysis, manuscript drafting; Churnosov M.I. – manuscript editing.
Conflicts of interest: The authors have no conflicts of interest to declare.
Funding: There was no funding for this study.
For citation: Alali O.M., Churnosov M.I. Genome-wide studies of uterine leiomyomas.
Akusherstvo i Ginekologiya/Obstetrics and Gynecology. 2023; (7): 28-38 (in Russian)
https://dx.doi.org/10.18565/aig.2023.156
Keywords
uterine leiomyomas
polymorphisms
genome-wide association studies (GWAS)
association
exon sequencing
candidate genes
replication study
Uterine fibroids (also known as leiomyomas, ULs, or myomas) are the most common female reproductive system tumors [1, 2]. They are benign monoclonal uterine smooth muscle tumors that develop from the myometrium [3, 4]. More than 70% of women have uterine fibroids by the time they reach menopause. They affect 20–40% of reproductive-age women [4] (they frequently regress after menopause), with a lifetime incidence of 30–70% [5]. Similar prevalence rates of uterine fibroids are also typical for women in the Russian Federation [6]. While many women with uterine fibroids are unaware of their symptoms, around a quarter of them do [7] and potential reproductive disruption, as well as other signs of fibroids' overall impact on health-related quality of life. Complications occur in 10% to 40% of pregnancies with UL, and miscarriage is up to twice as common in women with symptomatic UL [8].
Uterine fibroids are a major cause of gynecologic morbidity, one of the most prevalent reasons for gynecologic hospitalizations, and the most common reason for hysterectomy in the United States [9]. In the Russian Federation, uterine leiomyoma is the cause of hysterectomy in 50–70% of cases [10]. The yearly cost of treating this condition in the United States is projected to be over $34 billion, which is higher than the cost of treating breast and colon cancers combined [11]. Furthermore, fibroids-related infertility and pregnancy problems might increase overall costs and morbidity [9]. ULs have a significant morbidity incidence and can affect daily activities, relationships, and work performance [12]. Women with ULs have a much worse quality of life, which worsens as the number and severity of symptoms increase.
Uterine leiomyoma is a complex disease caused by the combination of several demographic, dietary, and hormonal risk factors [9, 13–15], as well as biological, epigenetic, and genetic causes [16, 17], with the genetic component accounting for 40 to 50% [18]. A considerable number of academics and scientists are now investigating the genetic basis for the formation, development, and progression of uterine leiomyomas.
No country in the world uses evidence on the effect of mononucleotide polymorphism genotypes on the risk of leiomyoma development or recurrence, although predicting such a risk could help guide clinical decision-making. The low odds ratios for all known polymorphic loci indirectly suggest that the genetic risk of leiomyoma development is usually determined by a constellation of several factors [15]. With this in mind, a reasonable approach to developing diagnostic systems is to use multiparametric methods for assessing genetic risk using genotyping across multiple loci simultaneously. However, the only known work that uses this principle remains Bongadgi N.S. et al. (2012) [19]. Genetic determinants, each of which by itself carries a high risk of disease are described [20, 21], but such markers are extremely rare in the population and, as a result, make an insignificant contribution to the overall morbidity. In addition, known determinants of this type are confined to distinct ethnoterritorial groups, which does not allow their use in other populations.
The whole genome association search (GWAS) is used to investigate the role of genetic markers in the creation and progression of various multifactorial disorders, including uterine leiomyomas [22]. Various research teams are presently investigating the genetic basis of ULs based on GWAS. At the same time, the findings collected are confusing, sometimes conflicting, and have little repeatability in different groups of the world.
It should be noted that the population of Russia is characterized by significant ethno-territorial diversity, which determines pronounced differences in the genetic structure of various populations of Russia, which can have a significant impact on the results of associative studies of candidate genes with UL. And this factor must be taken into account when planning and conducting genetic and epidemiological studies of uterine fibroids in Russian populations.
The aim of this review was to investigate the published genome-wide studies of ULs in order to recognize GWAS-significant polymorphism loci linked with ULs.
Materials and methods
The search for publications was carried out in the electronic databases PubMed, PubMed Central, eLibrary, in the GWAS catalog for the period from 2011 to the present by the keywords: Uterine leiomyomas, fibroids, GWAS studies, candidate genes.
Results and discussion
At the time of writing (July 2022), the National Human Genome Research Institute (GWAS) catalog of genome-wide studies (https://www.ebi.ac.uk/gwas/) presents the findings of eight studies on Uterine leiomyomas (Uterine fibroids).
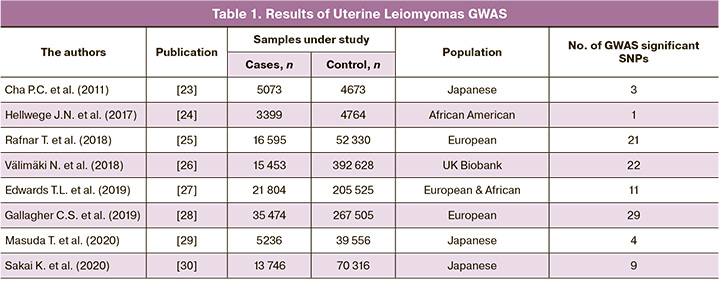
Cha P.C. et al. conducted the first GWAS research of uterine leiomyoma on a sample of Japanese women in 2011. A total of 457,044 single nucleotide polymorphisms (SNPs) were tested for their relationship with UL in 1,612 clinically confirmed cases (who showed severe symptoms of uterine fibroids, including dysmenorrhea or hypermenorrhea, or had a history of hysterectomy, myomectomy or uterine artery embolization) from the Japanese BioBank Project and 1,428 controls with no known UL history. The link was then replicated in an independent Japanese cohort of 3,466 cases and 3,245 controls with no known history of UL. Three loci on chromosomes, 10q24.33, 22q13.1, and 11p15.5, were shown to have genome-wide significant correlations with uterine fibroids. The SNPs showing the most significant association in a combination analysis at each of these loci were rs7913069 (p=8.65×10-14, odds ratio (OR)=1.47), rs12484776 (p=2.79×10-12, OR=1.23) and rs2280543 (p=3.82×10-12, OR=1.39), respectively (Table1) [23].
Hellwege J.N. et al. published the first multi-stage genome-wide association study (GWAS) of uterine fibroid risk in African American (AA) women in 2017 (675,206 SNPs have been studied). The associations with fibroid risk were modeled using logistic regression adjusted for main components, and the findings were meta-analyzed. Where observed a significant association among 3399 AA cases and 4764 AA controls (were confirmed by pelvic imaging, genotyped and imputed to 1000 Genomes) at rs739187 (risk-allele frequency=0.27) in CYTH4 (OR (95% confidence interval) = 1.23 (1.16–1.30), p-value=7.82×10-9). This first multi-stage GWAS for fibroids among AA women discovered a new fibroids risk loci within CYTH4 with probable biological significance for fibroids [24].
Rafnar T. et al. in 2018 published the first GWAS of leiomyoma in European populations (150,656 SNPs have been studied). The findings indicate a genetic connection between leiomyoma and other malignancies, as well as the importance of estrogen in tumor development. It conducted a meta-analysis of two genome-wide association studies of leiomyoma in European women (16,595 histologically confirmed leiomyoma cases and 52,330 controls). According to the findings of the meta-analysis, at least two genetic pathways are involved in the growth of leiomyomas, one linked to tumorigenesis (rs78378222 in TP53, rs10069690 in TERT, rs1800057 and rs1801516 in ATM, and rs7907606 at OBFC1) and the other linked to hormone-related traits at 1q36.12 (rs10917151 in CDC42/WNT4), 2p25.1 (rs10929757 and rs148143917 in GREB1), 20p12.3 (rs16991615 in MCM8), and 6q26.2 (rs58415480 in SYNE1/ESR1). The most significant association with leiomyoma is with a low-frequency 3’UTR variant in TP53, rs78378222 (p=4.03×10-37, meta-analysis of logistic regression, OR=1.74) [25].
Välimäki N. et al. conducted a genome-wide association research on 15,453 UL patients (were identified on the basis of both the self-reported UL phenotype and International Classification of Diseases (ICD) codes) and 392,628 controls in 2018 (805,426 SNPs have been studied). The genome-wide statistics revealed rs117245733, at 13q14.11, as the only SNP with a suggestive (p<1010-5) association in both the UKBB (OR=1.26; p=4.2×10-9) and Helsinki (OR=1.82; p=8.1×10-6) cohorts. Genes involved in genome stability were represented by TERT, TERC, OBFC1, TP53 and ATM. Genes involved in genitourinary development, WNT4, WT1, SALL1, MED12, ESR1, GREB1, FOXO1, DMRT1 and uterine stem cell marker antigen CD44, formed another strong subgroup. The combined risk from the 22 loci was linked to MED12 mutation-positive tumors [26].
Edwards T.L. et al. conducted in 2019, a two-stage meta-analysis of genetic variants in European and African ancestry women from seven electronic Medical Records and Genomics (eMERGE) network sites (3,704 imaging-confirmed cases and 5,591 imaging-confirmed controls) and women of African and European ancestry from UK Biobank (UKB, 5,772 cases and 61,457 controls) were included in the discovery genome-wide association study (GWAS) meta-analysis (827,762 SNPs have been studied). Final analysis with 21,804 cases and 205,525 controls discovered 326 genome-wide significant variants in 11 loci, including three novel loci at chromosome 1q24 (sentinel-SNP rs14361789; p=4.7×10-8), chromosome 16q12.1 (sentinel-SNP rs4785384; p=1.5×10-9) and chromosome 20q13.1 (sentinel-SNP rs6094982; p=2.6×10-8). Statistically significant findings further support previously identified loci such as SNPs near WT1, TNRC6B, SYNE1, BET1L, and CDC42/WNT4. It reports evidence of ancestry-specific findings for sentinel-SNP rs10917151 in the CDC42/WNT4 locus (p=1.76×10-24) [27].
Gallagher C.S. et al. conducted a GWAS meta-analysis in 35,474 cases (confirmed either based on self-report or clinically documented UL history) and 267,505 female controls of European ancestry in 2019. 302,979 SNPs have been studied in this GWAS, identifying eight novel genome-wide significant (p<5×10-8) loci associated with UL (2p23.2, 4q22.3, 6p21.31, 7q31.2,10p11.22, 11p14.1, 12q15, and 12q24.31), which include the following candidate genes of interest: HMGA1, BABAM2 and WNT2. In addition to verifying 21 previously reported loci, independent signals were found for ten of them. Heavy menstrual bleeding phenotypic stratification of UL in 3409 cases and 199,171 female controls showed genome-wide significant relationships at three of the 29 UL loci: 5p15.33 (TERT), 5q35.2 (FGFR4), and 11q22.3 (ATM). Four of the loci found in the meta-analysis are also linked to endometriosis risk; an epidemiological meta-analysis of 402,868 women reveals that those with a history of endometriosis are at least twice as likely to be diagnosed with UL [28].
Masuda T. et al. used data from the Biobank Japan Project to conduct genome-wide association studies of five gynecologic diseases in 2020 (5236 uterine fibroid, 645 endometriosis, 647 ovarian cancer, 909 uterine endometrial cancer, and 538 uterine cervical cancer, clinically diagnosed, and 39,556 shared female controls; (175,574 SNPs have been studied). Analyses performed under logistic model, linear mixed model, and model incorporating correlations identified four loci for uterine fibroids (rs7412010, chr:g.22436446 G>C at 1p36, CDC42/WNT4, p=1.2×10-12; rs12415148, chr10:g.105680586 T>C at 10q24, STN1 (OBFC1), p=3.5×10-10; rs12225799, chr11:g.241124 C>G at 11p15, BET1L/RIC8A, p=1.1×10-21; rs17332320, chr22:g.40711620 G>T at 22q31, TNRC6B, p=1.6×10-12). The direction of effect of previously described variations was concordant when the identical variants were included in these results [29].
Sakai K. et al. conducted a two-staged GWAS in 2020 with 13,746 UL cases (based on ultrasound examination and/or magnetic resonance imaging (MRI)) and 70,316 controls (without a history of UL and malignancies) from the Japanese population, followed by a replication study with 3,483 cases and 4,795 controls. 951,117 SNPs have been studied in this GWAS, found 9 significant loci, including a new one on 12q23.2 (rs17033114, p=6.12×10-25 with an OR=1.177 (1.141–1.213), LINC00485). Subgroup analysis pointed, 5 loci (3q26.2, 5p15.33, 10q24.33, 11p15.5, 13q14.11) were statistically significant among multiple leiomyomas and 2 loci (3q26.2, 10q24.33) were significant among submucous leiomyomas. Pleiotropic analysis revealed that all nine loci were linked to at least one proliferative illness, indicating that these genes play a part in the same neoplastic pathway. Furthermore, the rs2251795 (3q26.2) risk T allele was linked to greater telomere length in both normal and tumor tissues [30]. Beside this, there are contradictory data that both in karyotypically normal and abnormal samples of leiomyomas, telomeric repeats are on average ~40% shorter than in the corresponding myometrium [31]. The findings provide light on the important role of genetic causes in the etiology of leiomyoma.
Thus, for the period from 2011 to the present (2022), 8 GWAS ULs were performed (Table 1), as a result of which 34 genes and 79 GWAS-significant single nucleotide polymorphisms (SNPs) associated with the development of UL [23–30]. In Table 2 presented data on UL involved genes, showed associations with the disease in two or more GWAS (19 genes and 62 GWAS-significant polymorphisms were identified loci associated with the risk of developing UL).
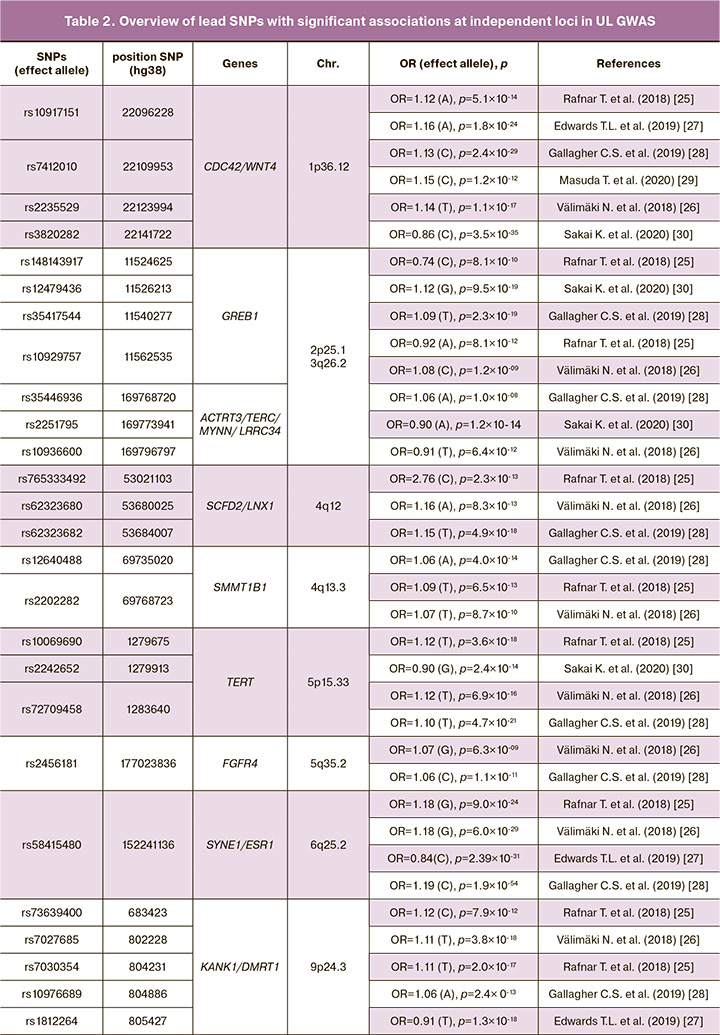
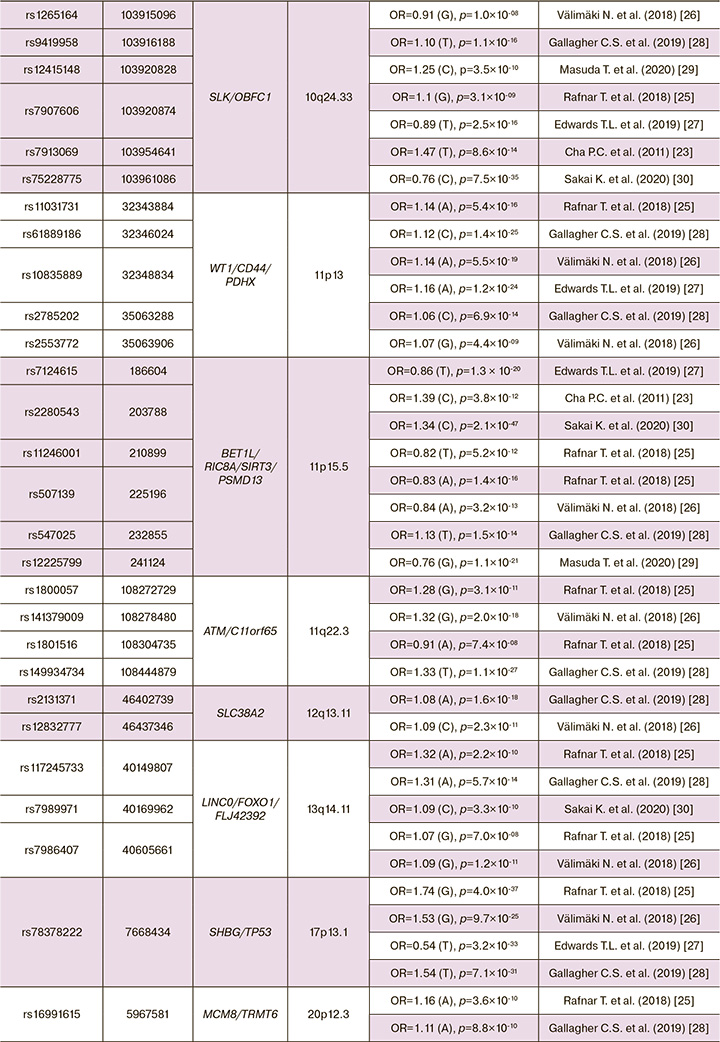
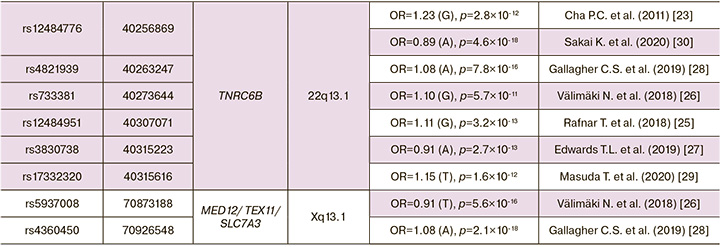
GWAS ULs were performed on residents of Japan (2 GWAS), Europe (3 GWAS) and others. Depending on published genome-wide association studies (GWAS) of uterine leiomyoma, and some common single nucleotide polymorphisms (SNPs) associated with leiomyoma risks, some genes and their nearby sites have been replicated in seven studies at: 10q24.3 (SLK/OBFC1) [23, 25–30], 11p15.5 (BET1L/RIC8A/SIRT3/PSMD13) [23, 25–30], 22q13.1 (TNRC6B) [23, 25–30], in six studies at: 1p36.12(CDC42/WNT4) [25–30], in four studies at: 2p25.1 (GREB1)[25, 26, 28, 30], 5p15.33 (TERT) [25, 26, 28, 30], 6q25.2 (SYNE1/ESR1)[25-28], 9p24.3 (KANK1/DMRT1) [25–28], 11p13 (WT1/CD44/PDHX)[25-28], 13q14.11 (LINC0/FOXO1 /FLJ42392) [25, 26, 28, 30], 17p13.1 (SHBG/TP53) [25–28], in three studies at: 3q26.2 (ACTRT3/TERC/MYNN/LRRC34)[26,28,30], 4q12 (SCFD2/LNX1) [25, 26, 28], 4q13.3 (SULT1B1) [25, 26, 28], 11q22.3 (ATM/C11orf65) [25, 26, 28], in tow studies at: 5q35.2 (FGFR4) [26, 28], 12q13.11 (SLC38A2) [26, 28], 20p12.3 (MCM8/TRMT6) [25, 28], Xq13.1 (MED12/TEX11/SLC7A3) [26, 28].
It should be noted that 15 genes out of 34 currently known GWAS significant genes showed association with the disease in only one GWAS, which is certainly not enough and dictates the need for further (replicative) studies of the role of these genes in the occurrence of the disease. In addition, uterine leiomyoma harbors genetic alteration of some driver genes including MED12 mutations, biallelic inactivation of FH and HMGA2 rearrangements [32, 33]. However, only a portion of the genetic variations of uterine leiomyoma could be explained.
The pathogenomics of UL offers fresh perspectives on how to prevent, diagnose, and treat this illness. For the treatment of fibroids with overexpression of the HMGA2 gene, inhibition of the signaling pathway linked to the activation of the insulin like growth factor 2 mRNA binding protein 2 (IGF2BP2) gene is particularly promising. A novel and promising approach to the pharmacotherapy of UL is the effective management of its size (suppression of the TGF- and ACVR1 genes). This is because ECM plays a critical role in the formation of UL. There are now more opportunities for determining the possibility of developing UL and the characteristics of its clinical manifestation thanks to the discovery of candidate genes for the disease as well as undesirable combinations of minor alleles and their epigenetic regulation. By doing this, one may also create innovative tactics for treating this important gynecological condition as well as tearly preventative strategies [34].
Single nucleotide polymorphisms (SNPs) have been also repeated in four studies as: rs58415480 [25-28] and rs78378222 [25-28]. And most of them have been repeated in two studies, such as: rs10917151 [25, 27], rs7412010 [28, 29], rs10929757 [25, 26], rs2202282 [25, 26], rs72709458 [26, 28], rs2456181 [26, 28], rs7907606 [25, 27], rs10835889 [26, 28], rs2280543 [23, 30], rs507139 [25, 26], rs117245733 [25, 28], rs7986407 [25, 26], rs16991615 [25, 28], rs12484776 [23, 30]. Thus, 16 SNPs from 79 GWAS-significant loci were replicated in genome-wide studies, which may indicate the reliability of data on the association of these polymorphisms with uterine myoma. Also, these 16 GWAS polymorphic loci replicated for UL can be recommended for further replication studies in other populations. The probability of confirming their connection with the disease in various ethno-territorial groups of the population is quite high. It is important to note that the vast majority of UL associated loci (63 out of 79 known to date) showed an association with the disease in only one GWAS and of course, additional (replicative) studies are needed for these loci, which will confirm (or disprove) their association with the disease.
Farther more exon sequencing has been performed in many researches along with GWAS. One study, which conducted by Yatsenko S.A. et al., used whole-exome sequencing and genome-wide arrays to investigate the genomic landscape of uterine leiomyomas. This research involved analyzing 16 recently frozen samples of leiomyoma and corresponding unaffected myometrium tissues, along with 153 leiomyosarcomas obtained from women who were diagnosed with uterine leiomyomas or leiomyosarcomas and had undergone abdominal hysterectomy as part of their clinical treatment. The authors identified several recurrent genetic alterations in genes involved in chromatin remodeling, cell proliferation, and cell signaling pathways. They also found that the genomic landscape of uterine leiomyomas is highly heterogeneous, MED12-negative leiomyomas include copy number changes affecting Mediator complex components such as MED8, MED18, CDK8, and long intergenic nonprotein coding RNA340 (CASC15), which may impact Mediator architecture and/or transcriptional activity. The authors found mutations, implicated in leiomyomagenesis, in COL4A6, DCN, and AHR genes, as well as novel genes NRG1, ADAM18, HUWE1, FBXW4, FBXL13, and CAPRIN1 [35].
In addition to study, which conducted by Kämpjärvi K. et al., used targeted exon sequencing to investigate the genetic mutations in uterine leiomyomas from a large cohort of women. 611 MED12 exon 2 mutation-negative tumors were analyzed for potential MED12 exon 1 mutations. The authors found that the majority of uterine leiomyomas had mutations in the mediator complex subunit 12 (MED12) gene, which is involved in transcriptional regulation, as five mutations were observed, all of which were in-frame insertion/deletions. According to transcriptome-wide expression data, MED12 exon 1 and exon 2 mutations result in the same distinct global gene expression pattern, with RAD51B being the most elevated gene. Exon 1 and exon 2 mutations impair the connection between MED12 and Cyclin C and CDK8/19 and remove the mediator-associated CDK kinase activity, according to immunoprecipitation and kinase activity experiments. The findings underscore the importance of MED12 in uterine leiomyomas, demonstrate that exon 1 and exon 2 have similar tumorigenic effects, and emphasize the need of include exon 1 in future MED12 screens [36].
Other study, which conducted by Firdaus R. et al., used targeted exon sequencing to investigate the genetic mutations in uterine leiomyomas from a cohort of German women. In this study, 22 multiple fibroids from both the same uterus and from different uteri of 4 women were tested for somatic mutations in MED12 hotspot regions. The authors found that the majority of uterine leiomyomas had mutations in the mediator complex subunit 12 (MED12) gene, which is involved in transcriptional regulation. Specifically, they identified multiple mutations in exon 2 of the MED12 gene, which were associated with a higher rate of leiomyoma growth, and flanking intronic regions, seven exonic variants and five intronic variants which provide evidence that multiple UL in the same uterus may not be clonal in origin. The study also compared the mutation profiles of uterine leiomyomas from German women to those from other populations, and found that the mutation patterns were similar across different ethnic groups [37].
This study, which conducted by Ajabnoor G.M. et al., used targeted exon sequencing to investigate the genetic mutations in uterine leiomyomas from a cohort of Saudi Arabian women. A total of 154 fresh tissue samples (consisting of leiomyoma and matching myometrial tissue) corresponding to 77 hysterectomized uterine specimens were collected at the Department of Histopathology, KAUH, Jeddah. The authors found that the majority of uterine leiomyomas had mutations in the mediator complex subunit 12 (MED12) gene, which is involved in transcriptional regulation. They observed that more than 44% (34/77) of Arab women's leiomyomas have a variety of MED12 mutations (30 missense, 1 splice site, and 3 indels). They discovered new somatic mutations in codons 36, 38, and 55, in addition to the known codon 44. The majority of genetically modified tumors (27/30; 90%) showed only one kind of genetic alteration, demonstrating that even a single allele shift in MED12 can have a significant influence on the transformation of normal uterine myometrium to leiomyomas. When the tumor is positive for the MED12 mutation (p<0.05), an intriguing inverse association between tumor size and LH is seen. The computational studies reveal that amino acid substitution mutations in the MED12 exon-2 area may lead to possible phenotypic and protein stability changes. The study also compared the mutation profiles of uterine leiomyomas from Saudi Arabian women to those from other populations, and found that the mutation patterns were similar across different ethnic groups. However, the authors noted that the frequency and distribution of MED12 mutations varied between different studies and populations [38].
This study, which conducted by Bertsch E. et al., used targeted exon sequencing to investigate the genetic mutations in uterine leiomyomas and leiomyosarcomas. A total of 178 patients with a histological diagnosis of conventional leiomyoma and 32 with uterine leiomyosarcoma were selected for study. They discovered that MED12 mutations were present in 74.7% (133/178) of leiomyomas, which was consistent with many independent researches. In comparison, MED12 mutations were found in just 9.7% (3/32) of leiomyosarcomas. These findings show that MED12 may have a functional role in both benign and malignant uterine smooth muscle tumors. The particular mutations found varied each tumor, however the majority of MED12 mutations were found in exon 2 of the MED12 gene. When HMGA2 expression in all leiomyomas and leiomyosarcomas was studied further, it was shown that HMGA2 overexpression was solely present in leiomyomas with no MED12 mutation, accounting for 10.1% (18/178) of total leiomyomas and 40% (18/45) of non-MED12 mutant leiomyomas. HMGA2 overexpression was observed in 25% (8/32) of leiomyosarcomas, and no MED12 mutations were discovered in HMGA2 positive leiomyosarcomas. These data clearly imply that MED12 mutations and HMGA2 overexpression are independent genetic events in leiomyomas, and that they may play separate roles in the formation of uterine leiomyomas [39].
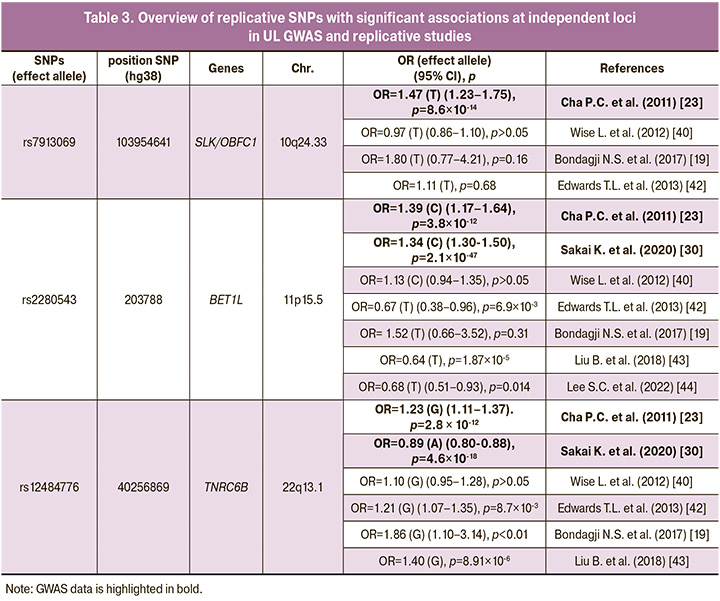
There are also conducted replicative GWAS studies of significant loci associated with development of uterine leiomyoma. The work of Wise L. et al. (2012) [40] is devoted to the study of genetic factors of uterine leiomyoma in African American women (genotyping of more than 1500 ethnically significant genetic markers was carried out). In a sample of 2453 patients confirmed by ultrasound or surgery and 2102 individuals in the control group (Black Women’s Health Study (BWHS)) involvement in the formation of uterine leiomyoma of three polymorphic loci of individual regions of three chromosomes 2q37 (rs7573626), 4p16.1 (rs9715724) and 10q26 (rs7100028) was revealed. It should be noted that associations of the three polymorphic loci with the development of the disease, previously shown in the GWAS work by Cha P.C. et al. (2011) in the Japanese population [23], were not confirmed in this study [40].
A replicative study of three GWAS-significant polymorphic loci for UL (data from Cha P.C. et al. (2011) in the Japanese population [23]) was carried out by Edwards T.L. et al. (2013)[41]. In a sample of American women of Caucasian origin, including 1086 patients with UL (determined by pelvic imaging) and 1549 individuals in the control group (formed from two cohorts – Right from the Start (RFTS) and the BioVU DNA biobank), associations with the development of uterine leiomyoma 2 SNPs were established – rs12484776 of the TNRC6B gene and rs2280543 BET1L gene. The association of rs12484776 of the TNRC6B gene with the volume of myomatous nodes, and rs2280543 of the BET1L gene with the development of intramural myomatous nodes was shown Edwards T.L. et al. (2013) [42].
A study conducted on Saudi women by Bondagji N.S. et al. in 2017, where genotyped 105 UL patients diagnosed through bimanual examinations, ultrasonographic imaging, and histopathological confirmation of uterine biopsies (collected from uterine hysterectomies) and 112 healthy controls for five genetic polymorphisms, and was examined the strength of the correlation between genotype and allele frequencies with risk of developing UL. The results found that Saudi women with UL had a high prevalence of the G allele of rs12484776 compared to the control group (OR, 1.86; 95% confidence interval [CI]: 1.10–3.14; p< 0.01). The authors of the work showed that individuals with AG genotype for the rs12484776 polymorphism are at a 2.6-fold greater risk of developing UL compared to those with other genotypes (OR, 2.69; 95% confidence interval [CI]: 1.45–5.00; p< 0.001). Genotype distribution frequencies for rs7913069 and rs2280543 were not shown to elevate the disease risk (for all tests p>0.05) [19].
The study that conducted in 2018 by Liu B. et al. aimed to replicate two initial significant genetic factors, TNRC6B and BET1L, in a Han Chinese population. A total of 674 women with UL diagnosed by ultrasonography and 1,381 individuals were recruited, and 55 SNPs mapped to TNRC6B and BET1L were selected and genotyped in samples from these subjects. Associations between targeted SNPs and relevant clinical features of UL were analysed in case only samples. Two SNPs, rs2280543 from BET1L (χ2=18.3, OR=0.64, p=1.87×10-5) and rs12484776 from TNRC6B (χ2=19.7, OR=1.40, p=8.91×10-6), were identified as significantly associated with the disease status of UL. Rs2280543 was significantly associated with the number of fibroid nodes (p=0.0007), while rs12484776 was significantly associated with node size (χ2=54.88, p=3.44×10-11). Both SNPs were a significant eQTL for their genes [43].
The research by Lee S.C. et al. (2022) is the first to replicate the link between BET1L rs2280543 and UL in Taiwanese women. A total of 341 UL patients and 1656 controls were recruited for the study. They integrated the digitized data from the Taiwan Biobank (TWB) database to the participants' medical records (collected diagnosed diseases, genotypic, lifestyle and biochemical data) from the National Health Insurance Research Database (NHIRD). Participants with the BET1L rs2280543 CT/TT genotype had an OR=0.69 and a 95% CI: 0.51–0.93 when compared to those with the BET1L rs2280543 CC genotype (wildtype). The vegetarian diet and UL had no significant relationship: OR=1.09 and 95% CI: 0.77–1.55. The interaction test between rs2280543 and vegetarian diet, on the other hand, was significant (p=0.046). When compared to those with the CC genotype, vegetarians with the CT/TT genotype had a reduced incidence of UL: OR (95% CI) 0.15 (0.05–0.47). They discovered that the BET1L rs2280543 CT/TT genotype was linked to a decreased incidence of UL, particularly among vegetarians [44].
It should be noted that the variations in their results (associations confirmed in some studies but not in others) may be caused by the unique genetic structure of the studied populations (determined by their ethnic characteristics and other factors), the unique characteristics of how environmental risk factors (nutrition, etc.) affect these people and other factors. Additionally, replication research on uterine fibroids must continue in order to identify the loci that define a certain ethno-territorial group women(population)'s susceptibility to the disease's development.
The results of the above- mentioned replicative studies are summarized in the table 3. GWAS and replicative studies performed indicate the most important role in the UL development of polymorphisms rs2280543 gene BET1L (11p15.5), rs12484776 TNRC6B (22q13.1) (Table 3). The rs2280543 gene BET1L (11p15.5) polymorphic locus was GWAS significant in two studies [23, 30] and was confirmed to be associated with the disease in three replication studies [42–44]. At the same time, the C allele of rs2280543 had a risk value for the development of uterine fibroids, and the T allele of this polymorphism had a protective value (Table 3). In two replication studies, the association of this polymorphism with UL was not confirmed [19, 40]. The rs12484776 TNRC6B (22q13.1) polymorphism has also been associated with uterine fibroids in two genome-wide studies [23, 30] and has been replicated in three independent studies [19, 42, 43]. The G allele variant rs12484776 TNRC6B (22q13.1) was associated with an increased risk of developing UL, and the A allele had a protective value for the disease (Table 3). One study did not find statistically significant associations of this polymorphism with UL [40]. Locus rs7913069 SLK/OBFC1 (10q24.33), although it is GWAS significant for uterine fibroids [23], but none of the three replicative studies conducted did not confirm the association with the disease [19, 40, 42].
Conclusion
The major genome-wide studies of UL were examined, and GWAS-significant polymorphisms associated with UL were determined. The materials gathered on GWAS-significant loci can be utilized in the selection of polymorphisms in replicative researches of UL in various populations, as well as to deepen the understanding of the molecular genetic mechanisms of the disease development. As a result of exon sequencing, the involvement of a number of genes (for example, MED12, etc.) in the formation of uterine fibroids has been shown, which significantly expands the understanding of the genetic determinants of the disease.
References
- Donnez J., Dolmans M.M. Uterine fibroid management: from the present to the future. Hum. Reprod. Update. 2016; 22(6): 665-86. https://dx.doi.org/10.1093/humupd/dmw023.
- Drayer S.M., Catherino W.H. Prevalence, morbidity, and current medical management of uterine leiomyomas. Int. J. Gynecol. Obstet. 2015; 131(2): 117-22. https://dx.doi.org/10.1016/j.ijgo.2015.04.051.
- Bulun S.E. Uterine fibroids. N. Engl. J. Med. 2013; 369(14): 1344-55.https://dx.doi.org/10.1056/NEJMra1209993.
- Ponomarenko I., Reshetnikov E., Polonikov A., Verzilina I., Sorokina I., Yermachenko A. et al. Candidate genes for age at menarche are associated with uterine leiomyoma. Front. Genet. 2021; 11: 512940.https://dx.doi.org/10.3389/fgene.2020.512940. 11:512940.
- Doherty L., Mutlu L., Sinclair D., Taylor H. Uterine fibroids: clinical manifestations and contemporary management. Reprod. Sci. 2014; 21(9):1067-92. https://dx.doi.org/10.1177/1933719114533728.
- Министерство здравоохранения Российской Федерации. Миома матки. Клинические рекомендации. 2020. [Ministry of Health of the Russian Federation. Uterine fibroids. Clinical guidelines. 2020. (in Russian)]. Available at: https://base.garant.ru/400455113/
- Alali O.M., Churnosov M.I. The etiopathogenesis of uterine leiomyomas: a review. Gynecology. 2023; 25(1): 22-30. https://dx.doi.org/10.26442/20795696.2023.1.201827.
- Van Heertum K., Barmat L. Uterine fibroids associated with infertility. Women’s Health. 2014; 10(6): 645-53. https://dx.doi.org/10.2217/WHE.14.27.
- Wise L.A., Laughlin-Tommaso S.K. Epidemiology of uterine fibroids–from menarche to menopause. Clin. Obstet. Gynecol. 2016; 59(1): 2-24.https://dx.doi.org/10.1097/GRF.0000000000000164.
- Адамян Л.В., ред. Миома матки: диагностика, лечение и реабилитация. Клинические рекомендации по ведению больных. М.: Научный центр акушерства, гинекологии и перинатологии имени академикаВ.И. Кулакова; 2015. 100с. [Adamyan L.V., ed. Uterine fibroids: diagnosis, treatment and rehabilitation. Clinical guidelines for managing patients. Moscow: Research Center of Obstetrics, Gynecology and Perinatology named after Academician V.I. Kulakov; 2015. 100p. (in Russian)].
- Alsudairi H.N., Alrasheed A.T., Dvornyk V. Estrogens and uterine fibroids: an integrated view. Res. Results Biomed. 2021; 7(2): 156-63.https://dx.doi.org/10.18413/2658-6533-2021-7-2-0-6.
- Al-Hendy A., Myers E.R., Stewart E. Uterine fibroids: burden and unmet medical need. Semin. Reprod. Med. 2017; 35(6): 473-80.https://dx.doi.org/10.1055/s-0037-1607264.
- Pavone D., Clemenza S., Sorbi F., Fambrini M., Petraglia F. Epidemiology and risk factors of uterine fibroids. Best Pract. Res. Clin. Obstet. Gynaecol. 2018; 46: 3-11. https://dx.doi.org/10.1016/j.bpobgyn.2017.09.004.
- Ciebiera M., Włodarczyk M., Słabuszewska-Jóźwiak A., Nowicka G., Jakiel G. Influence of vitamin D and transforming growth factor β3 serum concentrations, obesity, and family history on the risk for uterine fibroids. Fertil. Steril. 2016; 106(7): 1787-92. https://dx.doi.org/10.1016/j.fertnstert.2016.09.007.
- Пономаренко И.В., Чурносов М.И. Современные представления об этиопатогенезе и факторах риска лейомиомы матки. Акушерство и гинекология. 2018; 8: 27-32. [Ponomarenko I.V., Churnosov M.I. Current views on the etiopathogenesis and risk factors of uterine leiomyoma. Obstetrics and Gynecology. 2018; (8): 27-32 (in Russian)]. https://dx.doi.org/10.18565/aig.2018.8.27-32.
- Qin H., Lin Z., Vásquez E., Xu L. The association between chronic psychological stress and uterine fibroids risk: a meta‐analysis of observational studies. Stress Health. 2019; 35(5): 585-94. https://dx.doi.org/10.1002/smi.2895.
- Пономаренко И.В., Полоников А.В., Чурносов М.И. Полиморфные локусы гена LHCGR ассоциированы с развитием миомы матки. Акушерство и гинекология. 2018; 10: 86-91. [Ponomarenko I.V., Polonikov A.V. Polymorphic loci of the LHCGR gene are associated with the development of uterine leiomyma. Obstetrics and Gynecology. 2018; (10): 86-91.(in Russian)]. https://dx.doi.org/10.18565/aig.2018.10.86-91.
- Meadows K.L., Andrews D.M., Xu Z., Carswell G.K., Laughlin S.K., Baird D.D., Taylor J.A. Genome-wide analysis of loss of heterozygosity and copy number amplification in uterine leiomyomas using the 100K single nucleotide polymorphism array. Exp. Mol. Pathol. 2011; 91(1): 434-9.https://dx.doi.org/10.1016/j.yexmp.2011.03.007.
- Bondagji N.S., Morad F.A., Al‐Nefaei A.A., Khan I.A., Elango R., Abdullah L.S. et al. Replication of ПГАА loci revealed the moderate effect of TNRC6B locus on susceptibility of Saudi women to develop uterine leiomyomas. J. Obstet. Gynaecol. Res. 2017; 43(2): 330-8. https://dx.doi.org/10.1111/jog.13217.
- Nikpey P., Nazari T., Khalili S., Ebrahimi A. The role of epidermal growth factor receptor (EGFR) common gene mutations in Iranian women with uterine fibroids. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018; 229: 103-7.https://dx.doi.org/10.1016/j.ejogrb.2018.08.017.
- Zhang K., Winer H., Aissani B. Admixture mapping of genetic variants for uterine fibroids. J. Hum. Genet. 2015; 60(9): 533-8. https://dx.doi.org/10.1038/jhg.2015.60.
- Dehghan A. Genome-wide association studies. Methods Mol. Biol. 2018; 1793: 37-49. https://dx.doi.org/10.1007/978-1-4939-7868-7_4.
- Cha P.C., Takahashi A., Hosono N., Low S.K., Kamatani N., Kubo M., Nakamura Y. A genome-wide association study identifies three loci associated with susceptibility to uterine fibroids. Nat. Genet. 2011; 43(5): 447-50.https://dx.doi.org/10.1038/ng.805.
- Hellwege J.N., Jeff J.M., Wise L.A., Gallagher C.S., Wellons M., Hartmann K.E. et al. A multi-stage genome-wide association study of uterine fibroids in African Americans. Hum. Genet. 2017; 136(10): 1363-73. https://dx.doi.org/10.1007/s00439-017-1836-1.
- Rafnar T., Gunnarsson B., Stefansson O.A., Sulem P., Ingason A., Frigge M.L. et al. Variants associating with uterine leiomyoma highlight genetic background shared by various cancers and hormone-related traits. Nat. Commun. 2018; 9(1): 1-9. https://dx.doi.org/10.1038/s41467-018-05428-6.
- Välimäki N., Kuisma H., Pasanen A., Heikinheimo O., Sjöberg J., Bützow R. et al. Genetic predisposition to uterine leiomyoma is determined by loci for genitourinary development and genome stability. Elife. 2018; 7: e37110.https://dx.doi.org/10.7554/eLife.37110.
- Edwards T.L., Giri A., Hellwege J.N., Hartmann K.E., Stewart E.A., Jeff J.M. et al. A trans-ethnic genome-wide association study of uterine fibroids. Front. Genet. 2019; 10: 511. https://dx.doi.org/10.3389/fgene.2019.00511.
- Gallagher C.S., Mäkinen N., Harris H.R., Rahmioglu N., Uimari O., Cook J.P. et al. Genome-wide association and epidemiological analyses reveal common genetic origins between uterine leiomyomata and endometriosis. Nat. Commun. 2019; 10(1): 4857. https://dx.doi.org/10.1038/s41467-019-12536-4.
- Masuda T., Low S.K., Akiyama M., Hirata M., Ueda Y., Matsuda K. et al. GWAS of five gynecologic diseases and cross-trait analysis in Japanese. European J. Hum. Genet. 2020; 28(1): 95-107. https://dx.doi.org/10.1038/s41431-019-0495-1.
- Sakai K., Tanikawa C., Hirasawa A., Chiyoda T., Yamagami W., Kataoka F. et al. Identification of a novel uterine leiomyoma GWAS locus in a Japanese population. Sci. Rep. 2020; 10(1): 1197. https://dx.doi.org/10.1038/s41598-020-58066-8.
- Koltsova A.S., Efimova O.A., Malysheva O.V., Osinovskaya N.S., Liehr T., Al-Rikabi A. et al. Cytogenomic profile of uterine leiomyoma: in vivo vs. in vitro comparison. Biomedicines. 2021; 9(12): 1777. https://dx.doi.org/10.3390/biomedicines9121777.
- Mäkinen N., Mehine M., Tolvanen J., Kaasinen E., Li Y., Lehtonen H.J. et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science. 2011; 334(6053): 252-5.https://dx.doi.org/10.1126/science.1208930.
- Fusco A., Fedele M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer. 2007; 7(12): 899-910. https://dx.doi.org/10.1038/nrc2271.
- Baranov V.S., Osinovskaya N.S., Yarmolinskaya M.I. Pathogenomics of uterine fibroids development. Int. J. Mol. Sci. 2019; 20(24): 6151.https://dx.doi.org/10.3390/ijms20246151.
- Yatsenko S.A., Mittal P., Wood-Trageser M.A., Jones M.W., Surti U., Edwards R.P. et al. Highly heterogeneous genomic landscape of uterine leiomyomas by whole exome sequencing and genome-wide arrays. Fertil. Steril. 2017; 107(2): 457-66. https://dx.doi.org/10.1016/j.fertnstert.2016.10.035.
- Kämpjärvi K., Park M.J., Mehine M., Kim N.H., Clark A.D., Bützow R. et al. Mutations in exon 1 highlight the role of MED 12 in uterine leiomyomas. Hum. Mutat. 2014; 35(9): 1136-41. https://dx.doi.org/10.1002/humu.22612.
- Firdaus R., Agrawal P., Anagani M., Vijayalakshmi K., Hasan Q. Multiple mutations in exon-2 of med12 identified in uterine leiomyomata. J. Reprod. Infertil. 2021; 22(3): 201-9. https://dx.doi.org/10.18502/jri.v22i3.6720.
- Ajabnoor G.M., Mohammed N.A., Banaganapalli B., Abdullah L.S., Bondagji O.N., Mansouri N. et al. Expanded somatic mutation spectrum of MED12 gene in uterine leiomyomas of Saudi Arabian women. Front. Genet. 2018; 9: 552. https://dx.doi.org/10.3389/fgene.2018.00552.
- Bertsch E., Qiang W., Zhang Q., Espona-Fiedler M., Druschitz S., Liu Y. et al. MED12 and HMGA2 mutations: two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod. Pathol. 2014; 27(8): 1144-53.https://dx.doi.org/10.1038/modpathol.2013.243.
- Wise L.A., Ruiz-Narvaez E.A., Palmer J.R., Cozier Y.C., Tandon A., Patterson N. et al. African ancestry and genetic risk for uterine leiomyomata. Am. J. Epidemiol. 2012; 176(12): 1159-68. https://dx.doi.org/10.1093/aje/kws276.
- Edwards T.L., Michels K.A., Hartmann K.E., Velez Edwards D.R. BET1L and TNRC6B associate with uterine fibroid risk among European Americans. Hum. Genet. 2013; 132(8): 943-53. https://dx.doi.org/10.1007/s00439-013-1306-3.
- Edwards T.L., Hartmann K.E., Velez Edwards D.R. Variants in BET1L and TNRC6B associate with increasing fibroid volume and fibroid type among European Americans. Hum. Genet. 2013; 132(12): 1361-9.https://dx.doi.org/10.1007/s00439-013-1340-1.
- Liu B., Wang T., Jiang J., Li M., Ma W., Wu H., Zhou Q. Association of BET1L and TNRC6B with uterine leiomyoma risk and its relevant clinical features in Han Chinese population. Sci. Rep. 2018; 8(1): 7401.https://dx.doi.org/10.1038/s41598-018-25792-z.
- Lee S.C., Chou Y.H., Tantoh D.M., Hsu S.Y., Nfor O.N., Tyan Y.S., Liaw Y.P. Risk of uterine leiomyoma based on BET1L rs2280543 single nucleotide polymorphism and vegetarian diet. BMC Women's Health. 2022; 22(1): 139. https://dx.doi.org/10.1186/s12905-022-01721-1.
Received 18.04.2023
Accepted 30.06.2023
About the Authors
Ola Mohamad Alali, PhD student, Belgorod State National Research University, Belgorod, Russia, alaliola9@gmail.com, https://orcid.org/0000-0003-4370-6719Mikhail I. Churnosov, Dr. Med. Sci., Professor, Belgorod State National Research University, Belgorod, Russia, churnosov@bsu.edu.ru, https://orcid.org/0000-0003-1254-6134
Similar Articles

