

FMR1 inherited from women with premature ovarian failure: case series
Background: In recent years, the potential of transmission of an abnormal number of CGG triplet repeats in the FMR1 gene to the next generation as a biomarker for inheritance of early ovarian ageing has been of particular clinical interest.Rshtuni S.D., Zaretskaya N.V., Kuznetsova M.V., Marchenko L.A.
Objective: Analysis of health characteristics in children born to mothers with premature ovarian failure (POF), who had an abnormal number of trinucleotide CGG repeats in the FMR1 gene.
Materials and methods: A prospective, single-center, non-consecutive case series study included 90 women aged 18 to 39 years (the mean age was 33,5 years) with POF who underwent FMR1 gene testing to identify CGG repeats in FMR1 gene. The second stage of the study was quantitative assessment of the number of CGG trinucleotide repeats in 27 children born to the carriers of an abnormal number of triplet CGG repeats in the FMR1 gene.
Results: In 66.7% of cases, various FMR1 disorders were detected in offspring. In 7.4% of cases (2/27), adverse outcomes of inheritance of trinucleotide repeat expansion from premutation allele carriers were observed resulting in formation of Martin–Bell syndrome in sons. Stable inheritance of the FMR1 premutation was observed in 22.2% of cases (6/27), while severe phenotype of POF with early onset of the disease was observed in girls versus their mothers.
Conclusion: It is advisable that FMR1 premutation allele carriers should get genetic counseling that could help them to make decision about the use of assisted reproductive techniques (ART) to achieve pregnancy with donor oocytes or embryos.
Authors' contributions: Rshtuni S.D. – literature review; Marchenko L.A., Rshtuni S.D. – the concept and design of the study, writing the text of the article; Kuznetsova M.V., Rshtuni S.D. – material collection and processing; Zaretskaya N.V., Kuznetsova M.V. – article editing.
Conflicts of interest: The authors declare that they have no conflicts of interest.
Funding: The study was carried out without any sponsorship.
Ethical Approval: The study was approved by the local Ethics Committee of the Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of the Russian Federation (Registration No.5
of April 14, 2016).
Patients’ Consent for Publication: The patients have signed informed consent for publication of their data.
Authors' Data Sharing Statement: The data supporting the findings of this study are available on request from the corresponding author after approval from the principal investigator.
For citation: Rshtuni S.D., Zaretskaya N.V., Kuznetsova M.V., Marchenko L.A. FMR1 inherited from women with premature ovarian failure: case series.
Akusherstvo i Gynecologia/Obstetrics and Gynecology. 2023; (3): 91-98 (in Russian)
https://dx.doi.org/10.18565/aig.2022.285
Keywords
FMR1
CGG repeats
FMR1 premutation
Martin–Bell syndrome
premature ovarian failure (POF)
Premature ovarian failure (POF) is a multifactorial pathology, and genetic disorders play an important role in its etiopathogenesis [1, 2]. Monogenic forms of POF are inherited in accordance with Mendel's law. At the same time, in most cases, there is autosomal recessive inheritance with a low risk of recurrence in offsprings. Currently, more than 20 monogenic forms of POF are known, which are registered in the international medical database of human genes and genetic disorders (Online Mendelian Inheritance in Man (OMIM). Among them, the leading role belongs to the FMR1 gene [3]. It is known that X-linked recessive forms of the POF syndrome are most important for prediction of the prognosis in offspring. The FMR1 (fragile mental retardation) gene, a gene for mental retardation, is mapped on the long arm of the X chromosome at the Xq27.3 locus. Trinucleotide repeats are located in the 5' untranslated regions in exon 1 [4, 5].
The FMR1 gene is expressed in the oocyte, granulosa and luteal cells, and also in neurons [5]. Insufficient FMRP (fragile X mental retardation protein) production due to FMR1 gene mutation underlies abnormalities of folliculogenesis, leading to anovulation. At the same time, impaired neurogenesis in the CNS results in cognitive impairment and mental retardation. Expansion of CGG trinucleotide repeats (more than 200) is accompanied by abnormal methylation and lack of FMR1 expression [6]. Trinucleotide repeat instability can lead to formation of several genetic syndromes – Premature Ovarian Failure 1 (POF1), Martin–Bell syndrome (Fragile X syndrome, also called FRAXA syndrome) and Fragile X-associated tremor/ataxia syndrome (FXTAS).
According to the Russian and International recommendations, determination of CGG repeat numbers in the gene FMR1 should be performed in all women with POF and their female relatives [7 ,8].
In the practice of gynecologists in the Russian Federation, the average range of FMR1 gene CGG repeat distribution is 28–36 trinucleotides [9]. According to the American College of Medical Genetics and Genomics (ACMG) standards, there are 3 variations of CGG repeat in the FMR1 gene – a full mutation (fragile X syndrome, more than 200 repeats), premutation (expansion between 55–199 CGG triplet repeats) and grey zone (the range of 45–54 CGG repeats) [10]. In addition to this classification, according to Gleicher N. et al., alleles fewer than 28 trinucleotides are regarded as short repeats, while the range within 37–44 repeats is considered as FMR1 CGG repeat length expansion [11].
FMR1 premutation frequency has pronounced sex differences in women in 1:180–1:259 cases, while in men in 1:230–1:810 cases [12]. In familial cases of premature ovarian failure, the prevalence is 11.5%, and 3.2% in sporadic cases [10, 13]. For the first time in 1994, Schwartz et al. demonstrated in their multicenter study that in FMR1 premutation carriers, menopause started at the age before 40 three times more often than in the control group [14]. In 2004, it was found that in patients with FMR1 triplet repeat expansion to premutation in the gene, there was statistically significant increase of the level of follicle-stimulating hormone (FSH), low levels of circulating inhibins A and B not corresponding to their age norm, in combination with diminished ovarian reserve. At the same time, POF occurs in FMR1 premutation carriers only in 16–25% of cases [15]. Over the past ten years, it has been proved by Gleicher N. et al. that POF occurs with short CGG repeats in the FMR1 gene, but also in the carriers of trinucleotide repeats within the grey zone [11]. Thus, currently, an abnormal number of CGG repeats in the FMR1 gene can be used as a biomarker for prediction of premature ovarian failure.
There are very few researches in literature devoted to analysis of formation features of reproductive, somatic and neurological status in children born to mothers with POF, who are the carriers of the abnormal number of CGG repeats in the FMR1 gene.
The purpose of the study was assessment of health characteristics in children born to mothers with premature ovarian failure (POF) taking into account inheritance of the abnormal number of CGG repeats in the FMR1 gene.
Materials and methods
A prospective, single-center, non-consecutive case series study was carried out in the Department of Gynecologic Endocrinology and the Institute of Reproductive Genetics of the National Medical Research Center for Obstetrics, Gynecology and Perinatology named after Academician V.I. Kulakov of the Ministry of Health of Russia. The study included 90 women with POF, who underwent medical tests and/or treatment in accordance with our clinical activity. The patients were recruited for the study from 2018 to 2021. POF was diagnosed on the basis of the Russian clinical recommendations [8]. The study was approved by the local Ethical Committee of the National Medical Research Center for Obstetrics, Gynecology and Perinatology named after Academician V.I. Kulakov of the Ministry of Health of Russia (registration No. 5 of April 14, 2016. Each patient signed informed consent for participation in the study in accordance with The Declaration of Helsinki. Among them, 19 patients signed informed consent for examination of their children.
Inclusion criteria in the study were: the established diagnosis of POF; the age of women at the time of diagnosis 18–39 years; 46XX karyotype. Exclusion ctiteria were: no children in women with POF. Non-inclusion criteria were primary or iatrogenic, hypergonadotropic, normogonadotropic and hypogonadotropic amenorrhea.
To verify the diagnosis of POF, the functional state of the hypothalamic-pituitary-ovarian system was assessed based on measuring the levels of follicle-stimulating hormone (FSH), estradiol (E2) in blood serum by the electrochemiluminescent method using the automatic immunochemical analyzer Cobas e411 (Roche Diagnostics GmbH, Germany). Serum Anti-Müllerian hormone (AMH) in blood serum was quantified via two-step sandwich enzyme immunoassay using the AMH Gen II ELISA kit (Beckman Coulter, USA).
Pelvic ultrasound was performed using ultrasound scanner 2000 Toshiba SSA-240 (Japan) with transvaginal convex probe frequency 7.5 MHz.
Karyotype was determined using the standard technique (G-banding pattern, Seabright). The method for determining the number of CGG repeats in the FMR1 gene using amplification of the promoter region of the gene, including the CGG region, was performed by PCR using fluorescently labeled primers and determining the exact length of PCR products by fragment analysis on analyzer Applied Biosystems (reagent kit “DNA-Technology”) in accordance with the Protocol “Standard operational procedures (SOP) for detection of the number of CGG repeats in the FMR1 gene”.
Results
The study included 90 women with POF. The mean age of patients was 33/5 years. The mean age of the onset of the disease was 31 years. The results of hormonal tests were: mean serum values of FSH – 61.88 mIU/mL, estradiol (E2) – 44.7 pmol/L., АМH – 0,13 ng/mL. Stage I of the study was determination of the number of CGG repeats in the FMR1 gene in all women with the established diagnosis of POF (Table 1).
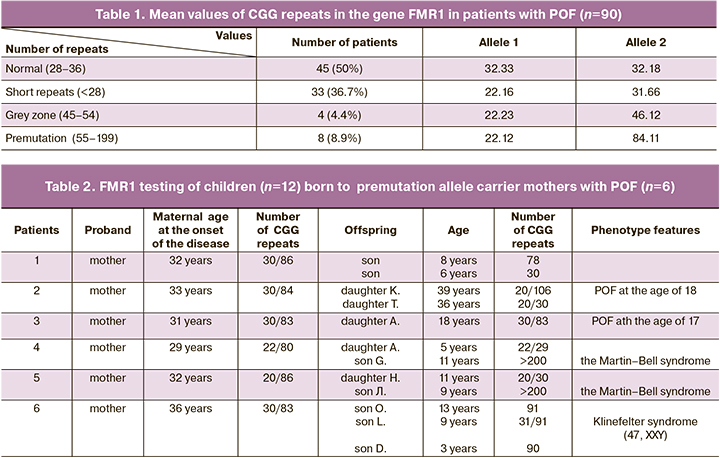
In 45/90 patients (50%), various changes in trinucleotide repeat number were identified with regard to normal number of repeats. At the same time, the FMR1 premutation was identified in 8/90 (8,9%) women, trinucleotide repeats in the grey zone were in 4/90 women (4.4%), and short repeats were in 33/90 patients (36.7%). In two cases, FMR1 premutation carriers had no children. Due to this fact, they were further excluded from the study. The second stage of the study was genetic testing of 27 children born to 19 carriers of alleles with abnormal number of CGG repeats.
In the first case series, inheritance of variations of trinucleotide repeats transferred from mothers with FMR1 premutation to their offsprings and their reproductive status were assessed. The results of FMR1 testing of premutation carrier mothers and their offsprings are shown in Table 2. In 6 premutation allele carriers, menarche occurred timely, and they had regular menstruation before the onset of the disease. Also, their reproductive function was preserved. This was confirmed by occurrence of 18 pregnancies, and 12 of them ended in birth of 7 boys and 5 girls. In the remaining 6 cases, medical abortions were performed on women’s request. Reproductive function was realized before the age of 30 (the mean age 27.6) in women included in this case series. The mean age of the onset of the disease was 32 years. Maternal allele 1 ranged from 20 to 30 triplet repeats, and allele 2 ranged from 80 to 86 triplet repeats.
The analysis of inheritance of the changes in the CGG repeat number in FMR1 gene in children born to permutation carriers showed that in a son (1/12 children, 8.3%) triplet repeats were in the normal range due to maternal allele 1 inheritance. In 3 daughters (3/12 children, 25 %) short repeats were identified. At the same time, allele 1 was inherited from their mothers, and allele 2 was probably inherited from their fathers. Detailed evaluation of anamnesis showed that puberty was in 2 girls, the third girl had timely menarche and until present, had regular menstrual cycle. In 2 girls and 4 boys (6/12. 50%), FMR1 premutation was inherited from their mothers. The number of CGG repeats ranged from 78 to 106. At the same time, their expansion during the study was observed in 4 children, reduction of CGG repeats was in 1 child, transmission in a stable manner was in one case. Thorough evaluation of the anamnesis identified that 1 boy went through puberty in accordance with his age. Three boys were of pubertal period. Additionally, numerical chromosome abnormalities were found in one of them (Klinefelter syndrome). Two daughters had a severe phenotype of POF with early onset of the disease at the age of 17 and 18, (4–5 years after menarche). It is notable that in their mothers, the onset of the disease was later (19 and 21 years after menarche). Expansion of trinucleotides in the FMR1 gene to full mutation was found in 2 sons (2/12, 16.7%). The boys were born with Martin–Bell syndrome and typical clinical manifestation (psychopathic and speech disorders, motor disinhibition, signs of autism) and phenotypic manifestation of the disease (dolichocephaly, protruding forehead, elongated face, massive chin, large size of auricles, epicanthus, large hands and feet, joint hypermobility, macroorchism).
In the second case series, inheritance variations of abnormal trinucleotide repeats in the grey zone in the FMR1 gene transferred from mothers to their offsprings and their reproductive status were evaluated. The results of the genetic testing of mothers with repeats in the grey zone and their offsprings are shown in Table 3. In all tested patients, who were carriers of triplet alleles in the grey zone, menarche occurred timely. They had regular menstruation before the onset of the disease. Their reproductive function was preserved. This was confirmed by occurrence of 7 pregnancies, 4 of them ended in birth of 2 boys and 2 girls. The mean age at the time of childbirth was 25.5. years. Evaluation distribution of triplet repeats in children showed that in 50% (2/4) it was within the range of population norm, and in 50% (2/4) there was inheritance of short trinucleotide repeats. All children born in this case series were of prepubertal period without diagnosed neurological and mental disorders.
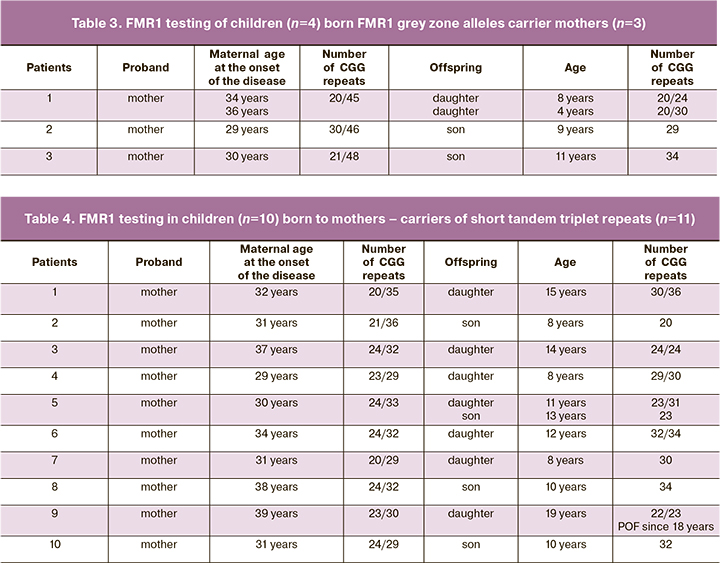
In the third case series inheritance variations of abnormal trinucleotide repeats with short repeats in the FMR1 gene transferred from mothers to their offsprings and their reproductive status were evaluated. The results of the genetic testing of mothers with short repeats in the FMR1 gene are shown in Table 4. In all tested patients, who were carriers of triplet alleles in the range of shorter repeats, menarche occurred timely. They had regular menstruation before the onset of the disease. Their reproductive function was preserved. This was confirmed by occurrence of 17 pregnancies, 11 of them ended in birth of 4 boys and 7 girls. Other pregnancies ended in medical abortions on women’s request. The mean age of patients at the time of childbirth was 29.7 years. Maternal allele 1 ranged from 20 to 40 CGG repeats, allele 2 ranged from 20 to 36 trinucleotides. Stable transmission of short repeats was identified in 3 daughters and 2 sons (5/11, 45.5%), in 4 girs and 2 boys (6/11, 54,5%). Triplet repeats were in the range of reference values. Out of 11 children, 7 were of prepuberal period, puberty in 3 children was according to their age. All children were without diagnoses of somatic and neurological disorders. One daughter aged 19 (1/11, 9.1%) was diagnosed with severe phenotype of POF at the age of 18, while in mother the onset of the disease was at 31.
Discussion
For many years, carriage of FMR1 premutation was not associated with development of somatic disorders. However, in the recent decades, it has been proved that comorbid pathology can develop in female premutation carriers, and in its structure, POF is in 16–25% of cases. At the same time, in addition to disorders of the reproductive system, autoimmune disorder, fragile X-associated tremor/ataxia syndrome, as well as neurocognitive impairment and emotionally unstable personality disorder can develop. In connection with the foregoing, the discussion of our results of evaluation of health characteristics of children born to mothers with POF who are carriers of the abnormal number of trinucleotide CGG repeats in the FMR1 gene is both of scientific and practical significance.
According to numerous literature sources, it has been proved that the risk of expansion of CGG repeats in the next generation to a full mutation increases with increasing number of trinucleotides in the maternal FMR1 gene. According to Nolin S.L. et al., during testing of 936 women with various changes in repeat number in the FMR1 gene in the premutation range, the risk of full mutation in the next generation was calculated taking into account maternal CGG repeat length. The risk was 3.7% in the range of 55–59 (n=26) CGG trinucleotide repeats, 57,8% in the range of 80–89 (n=133), 80% in the range of 90–99 (n=118). Maximum risk (94–100%) was observed with CGG repeat length in the range of 100–109 (n=26). Unfortunately, this study was carried out without estimation of ovarian reserve in these women and subsequent development of POF [16]. The data obtained by us showed that in mothers with POF, who had triplet repeats in the permutation range 22/80 and 20/86, expansion of CGG trinucleotides to a full mutation and birth of sons with Martin–Bell syndrome within one generation was diagnosed in 16.7% of cases, that is 3.7 times less compared to the number of repeats in the above study [16] (Figure).
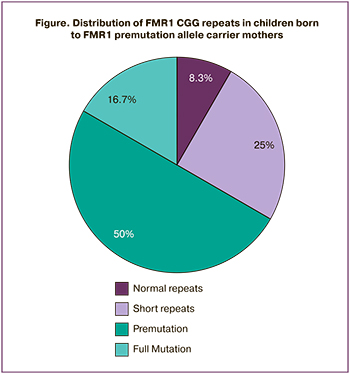
At the same time the frequency of formation of Martin-Bell syndrome was comparable to the data reported by Beke A. et al. They demonstrated that in 9 women with POF, who were FMR1 premutation carriers, Martin–Bell syndrome was diagnosed even less often, in 11.1% of cases [17].
At the end of the XXth century, it was found that normal CGG repeats were interrupted by AGG trinucleotides most often in the 10th and and 20th positions. This provides stability of their transmission. Lack or insufficient number of AGG interruptions leads to instability of CGG triplet repeats, expressed expansion and formation of Martin–Bell syndrome in the next generation [18]. However, unfortunately, we have not performed expanded analysis of the FMR1 gene with simultaneous identification of CGG and AGG trinucleotides in mothers with FMR1 premutation.
In our study, in 50% of cases (6/12) we observed that FMR1 premutation was inherited in a stable manner in children from women with POF. Two daughters with premutation had severe phenotype of POF. The onset of the diseases was at the age of 17 and 18 years. At the same time, in the study by Tabeeva G.I. et al., testing of 200 patients with POF showed that the mean age of the onset of the disease was 29.4±7.66 years [19].
In recent years, the role of short CGG repeats in the FMR1 gene associated with neurologic and reproductive system disorders is being actively discussed in published literature [11]. In our study, careers of short repeats were identified in 36.7% of cases. In 2014, Mailick M.R. et al. presented a unique long-term study, that included 10 317 persons, who were obseved during 54 years. By the end of the study, the age of respondents reached 72 years. At the final stage of the study, FMR1 CGG repeat analysis was performed was performed in 3469 (51.5%) women. As a result of analysis, 20.8% of carriers of short repeats were identified, that was 1.8 times less frequently versus our patients with POF. The authors noted that in female carriers of short CGG repeats in the FMR1 gene, breast cancer was diagnosed 2 times more frequently and endometrial cancer 4 times more frequently versus carriers of the normal range of CGG repeats. Their offsprings were diagnosed with severe mental retardation significantly more often compared to the population data (OR 1.68, р<0.05). At the same time, tremor/ataxia syndrome prevailed in male carriers of short repeats [20]. According to the data obtained by us, with stable transmission of short CGG repeats in 9.1% of cases, severe phenotype of POF with the onset of the disease at the age of 18 was diagnosed in daughter.
It is known that triplet repeat expansion within the grey zone can be transmitted to subsequent generations. However, in our study, due to limited sample size, this phenomenon was not confirmed [21, 22].
As a result of the study performed by us, it was proved that the variants of inheritance of CGG repeat number in children born to mothers with variations of abnormal CGG trinucleotides in the FMR1 gene are the following:
1) Normal triplet CGG repeats were found in 33.3% (9/27) of cases;
2) Short repeats were in 37% (10/27). With stable transmission of short repeats, severe phenotype of POF in daughter was identified in 9.1% of cases.
3) FMR1 premutation and formation of severe POF phenotype in daughter was found in 22.2% (6/27) of cases.
4) CGG repeat expansion to a full mutation and birth of boys with Martin–Bell syndrome was identified in 7.5% (2/27).
Currently, developments in techniques of molecular genetics make it possible planning of a biological child for female carriers of FMR1 premutation and heterozygous mutation. When discussing the reproductive prognosis, it is usually recommended to provide genetic counseling for women, where female patients with POF are informed about the age deadline for childbearing (conceiving before age 30 is preferable), high risk of giving birth to a boy with Martin–Bell syndrome, and possibilities of preimplantation genetic testing of embryos ( determination of the sex of the embryo and its selection in favor of female) [23]. In addition, the patients should be informed that in occurrence of spontaneous pregnancy, they should undergo prenatal diagnostic testing that includes: determination of the sex of the embryo at 10 weeks of pregnancy by non-invasive prenatal fetal DNA testing in maternal blood; diagnostic ultrasound of congenital fetal anomalies and fetal sex; invasive prenatal karyotype testing (including sex determination) and FMR1 mutation testing by chorion biopsy at 10–13 weeks of pregnancy, or amniocentesis at 16–20 weeks of pregnancy.
However, sex selection of embryo/fetus in favor of female does not always allow avoiding the birth of a girl with reproductive disorders inherited from her mother. A detailed study of the influence of AGG interruptions at FMR1 loci may be a resource for clarifying the origin of inherited trinucleotide repeats [24]. The ambiguous role of short repeats in the development of POF and risks for offspring requires further study. Considering other multiple monogenic forms of POF, it can be recommended to search mutations in other genes associated with POF using whole exome sequencing in female female grey zone carriers of repeats and atypical manifestations.
Limitations of the study: small sample size; no results of testing in fathers to determine CGG repeats in the FMR1 gene. No results of testing fathers for AGG interruptions in the CGG repeat locus.
Conclusion
Adverse outcomes in the offsprings were observed in case series due to maternal carriers of FMR1 premutation alleles. Usually, it was the ground to recommend these women to conceive using donor eggs or embryos in the programs of assisted reproductive technology. Thus, when this category of patients plan pregnancy, geneticists should take into consideration the exact number of CGG repeats in the FMR1 gene, as well as the number of AGG interruptions to obtain reliable results for individual risk assessment.
In our opinion, to detect variations of inheritance, it is necessary to make genealogical analysis of patients with POF, who are carriers of the abnormal number of CGG repeats in the FMR1 gene, taking into consideration the results of genetic testing of their first-degree and second-degree relatives. To detect phenotypic features in offsprings at the early stage, it is advisable to conduct counselling by multidisciplinary team of pediatrician, pediatric gynecologist, geneticist and neurologist, in order to recommend behavioral or drug therapy for children.
References
- Qin Y., Jiao X., Simpson J.L., Chen Z.-J. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum. Reprod. Update. 2015; 21(6): 787-808. https://dx.doi.org/10.1093/humupd/dmv036.
- França M.M., Mendonca B.B. Genetics of ovarian insufficiency and defects of folliculogenesis. Best Pract. Res. Clin. Endocrinol. Metab. 2022; 36(1): 101594. https://dx.doi.org/10.1016/j.beem.2021.101594.
- Bouilly J., Beau I., Barraud S., Bernard V., Azibi K., Fagart J. et al. Identification of multiple gene mutations accounts for a new genetic architecture of primary ovarian insufficiency. J. Clin. Endocr. Metab. 2016; 101(12): 4541-50.https://dx.doi.org/10.1210/jc.2016-2152.
- Tabolacci E., Nobile V., Pucci C., Chiurazzi P. Mechanisms of the FMR1 repeat instability: how does the CGG sequence expand? Int. J. Mol. Sci. 2022; 23(10): 5425. https://dx.doi.org/10.3390/ijms23105425.
- Eichler E.E., Richards S., Gibbs R.A., Nelson D.L. Fine structure of the human FMR1 gene. Hum. Mol. Genet. 1993; 2(8): 1147-53.https://dx.doi.org/10.1093/hmg/2.8.1147.
- Chen L.S., Tassone F., Sahota P., Hagerman P.J. The (CGG)n repeat element within the 5' untranslated region of the FMR1 message provides both positive and negative cis effects on in vivo translation of a downstream reporter. Hum. Mol. Genet. 2003; 12(23): 3067-74. https://dx.doi.org/10.1093/hmg/ddg331.
- Webber L., Davies M., Anderson R., Bartlett J., Braat D., Cartwright B. et al.; European Society for Human Reproduction and Embryology (ESHRE) Guideline Group. ESHRE guideline: management of women with premature ovarian insufficiency. Hum. Reprod. 2016; 31(5): 926-37.https://dx.doi.org/10.1093/humrep/dew027.
- Министерство здравоохранения Российской Федерации. Клинические рекомендации. Аменорея и олигоменорея. М.; 2021. [Ministry of Health of the Russian Federation. Clinical guidelines. Amenorrhea and oligomenorrhea. Moscow; 2021. (in Russian)].
- Шамилова Н.Н., Марченко Л.А., Долгушина Н.В., Кузнецова Е.Б., Залетаев Д.В. Роль генетических и аутоиммунных нарушений в развитии преждевременной недостаточности яичников. Акушерство и гинекология. 2012; 4-2: 67-72. [Shamilova N.N., Marchenko L.A., Dolgushina N.V., Kuznetsova E.B., Zaletayev D.V. Role of genetic and autoimmune disorders in the development of premature ovarian failure. Obstetrics and Gynecology. 2012;( 4-2): 67-72. (in Russian)].
- Spector E., Behlmann A., Kronquist K., Rose N.C., Lyon E., Reddi H.V.; ACMG Laboratory Quality Assurance Committee. Laboratory testing for fragile X, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021; 23(5): 799-812.https://dx.doi.org/10.1038/s41436-021-01115-y.
- Gleicher N., Barad D.H. The FMR1 gene as regulator of ovarian recruitment and ovarian reserve. Obstet. Gynecol. Surv. 2010; 65(8): 523-30.https://dx.doi.org/10.1097/OGX.0b013e3181f8bdda.
- Dombrowski C., Lévesque S., Morel M.L., Rouillard P., Morgan K., Rousseau F. Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum. Mol. Genet. 2002; 11(4): 371-8.https://dx.doi.org/10.1093/hmg/11.4.371.
- Bussani C., Papi L., Sestini R., Baldinotti F., Bucciantini S., Bruni V., Scarselli G. Premature ovarian failure and fragile X premutation: a study on 45 women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004; 11(2): 189-91.https://dx.doi.org/10.1016/j.ejogrb.2003.06.003.
- Schwartz C.E., Dean J., Howard-Peebles P.N., Bugge M., Mikkelsen M., Tommerup N. et al. Obstetrical and gynecological complications in fragile X carriers: a multicenter study. Am. J. Med. Genet. 1994; 51(4): 400-2.https://dx.doi.org/10.1002/ajmg.1320510419.
- Welt C.K., Smith P. C., Taylor A.E. Evidence of early ovarian aging in fragile X premutation carriers. J. Clin. Endocrinol. Metab. 2004; 89(9): 4569-74.https://dx.doi.org/10.1210/jc.2004-0347.
- Nolin S.L., Brown W.T., Glicksman A., Houck G.E. Jr, Gargano A.D., Sullivan A. et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 2003; 72(2): 454-64.https://dx.doi.org/10.1086/367713.
- Beke A., Piko H., Haltrich I., Karcagi V., Rigo J. Jr, Molnar M.J., Fekete G. Study of patterns of inheritance of premature ovarian failure syndrome carrying maternal and paternal premutations. BMC Med. Genet. 2018; 19(1): 113. https://dx.doi.org/10.1186/s12881-018-0634-5.
- Eichler E.E., Holden J.J., Popovich B.W., Reiss A.L., Snow K., Thibodeau S.N. et al. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 1994; 8(1): 88-94. https://dx.doi.org/10.1038/ng0994-88.
- Табеева Г.И., Шамилова Н.Н., Жахур Н.А., Позднякова А.А., Марченко Л.А. Преждевременная недостаточность яичников – загадка XXI века. Акушерство и гинекология. 2013; 12: 16-21. [Tabeeva G.I., Shamilova N.N., Zhakhur N.A., Pozdnyakova A.A. Marchenko L.A. Premature ovarian failure is a mystery of the 21st century. Obstetrics and Gynecology. 2013; (12): 16-21. (in Russian)].
- Mailick M.R., Hong J., Rathouz P., Baker M.W., Greenberg J.S., Smith L., Maenner M. Low - normal FMR1 CGG repeat length: phenotypic associations. Front. Genet. 2014; 5: 309. https://dx.doi.org/10.3389/fgene.2014.00309.
- Fernandez-Carvajal I., Lopez Posadas B., Pan R., Raske C., Hagerman P.J., Tassone F. Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J. Mol. Diagn. 2009; 11(4): 306-10. https://dx.doi.org/10.2353/jmoldx.2009.080174.
- Terracciano A., Pomponi M.G., Marino G.M., Chiurazzi P., Rinaldi M.M., Dobosz M., Neri G. Expansion to full mutation ofa FMR1 intermediate allele over two generations. Eur. J. Hum. Genet. 2004; 12(4): 333-6.https://dx.doi.org/10.1038/sj.ejhg.5201154.
- Rajan-Babu I.-S., Lian M., Cheah F.S.H., Chen M., Tan A.S.C., Prasath E.B., Loh S.F., Chong S.S. FMR1 CGG repeat expansion mutation detection and linked haplotype analysis for reliable and accurate preimplantation genetic diagnosis of fragile X syndrome. Expert Rev. Mol. Med. 2017; 19: e10.https://dx.doi.org/10.1017/erm.2017.10.
- Ardui S., Race V., de Ravel T., Van Esch H., Devriendt K., Matthijs G., Vermeesch J.R. Detecting AGG interruptions in females with a FMR1 premutation by long-read single-molecule sequencing: A 1 year clinical experience. Front. Genet. 2018; 9: 150. https://dx.doi.org/10.3389/fgene.2018.00150.
Received 30.11.2022
Accepted 23.03.2023
About the Authors
Sandra D. Rshtuni, postgraduate student, Department of Endocrinological Gynecology, V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, rshtunisandra@gmail.com, 117997, Russia, Moscow, Ac. Oparina str., 4.Nadezhda V. Zaretskaya, PhD, Head of the Laboratory of Clinical Genetics, V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, znadezda@yandex.ru, 117997, Russia, Moscow, Ac. Oparina str., 4.
Maria V. Kuznetsova, PhD (Bio), Senior Researcher at the Laboratory of Molecular Genetic Methods of the Institute of Reproductive Genetics, V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, mkarja@mail.ru, 117997, Russia, Moscow, Ac. Oparina str., 4.
Larisa A. Marchenko, Dr. Med. Sci., Professor, Department of Endocrinological Gynecology, V.I. Kulakov National Medical Research Center for Obstetrics,
Gynecology and Perinatology, Ministry of Health of Russia, l_marchenko@yandex.ru, 117997, Russia, Moscow, Ac. Oparina str., 4.
Similar Articles

