

Risk-based analysis of test panels for carrier screening for monogenic disorders
A number of autosomal recessive diseases with well-known genetic markers are relatively common in the Russian Federation. The diagnostic effectiveness of test panels is traditionally based on the prevalence of determinable mutations, which is rather difficult to detect in heterogeneous populations.Zobkova G.Yu., Donnikov A.E., Prytkov A.N., Demikova N.S., Ragimov A.A.
Objective: To evaluate the effectiveness of the screening panel for carriage of CFTR gene mutations in the Russian Federation using a risk-based approach.
Materials and methods: Molecular genetic testing of blood samples of 1000 healthy donors was carried out by analyzing the 24 most frequent mutations in the CFTR gene using the real-time PCR with the analysis of melting profile.
Results: The study found out 29 mutation carriers among 1000 healthy individuals. The total prevalence of detectable mutations was 2.9%. The vulnerability of the panel with a 95% probability is no more than 31%, which is close enough to the value of the vulnerability theoretically calculated basing on the prevalence of the mutations included (24.5). When using this panel (with the carriage of mutations in the CFTR gene), the risk of having an affected child in both partners will be only 1/170068, which is 17 times lower than the general population risk.
Conclusion: Use of this screening panel in both partners may reduce the risk of affected offspring by 17 times, compared to the general population risk. Using a risk-based approach allowed us to effectively evaluate the vulnerability of the panel. If the incidence of the disease is known, this technique is applicable for any heterogeneous population.
Keywords
hereditary diseases
preconceptional genetic screening
cystic fibrosis
genetic risk
carrier screening
risk management
Risk management in healthcare allows reducing the number of various unfavorable consequences. This includes not only risks associated with healthcare delivery, but also financial, organizational, legal and many other risks. When developing medical devices, manufacturers should apply the same principles (EEC Decision №27 of February 12, 2016). The term “risk-based thinking” (ISO 9001:2015) implies, first of all, risk identification, its description and quantitative evaluation. This study deals with efficacy evaluation of screening panels for detection of genetic markers by means of risk-based analysis.
A number of autosomal recessive diseases with well-known genetic markers are relatively common in the Russian Federation. The specific feature of autosomal recessive disorder is that a child receives defective variants of the key gene from both parents. Besides, more commonly both parents are intact heterozygous carriers of this genetic deficit, and in this case the possibility of affected offspring rises up to 25%. Timely detection of such couples gives opportunity to offer them various reproductive strategies in order to have healthy offspring in future; therefore preconception genetic testing for heterozygous carriage of autosomal recessive disorders is an effective tool for primary prevention.
One of the main limiting factors for large-scale implementation of preconception genetic testing is its high cost. Considerable cost saving can be achieved by focusing on the most frequent mutations, typical for a selected population. It is obvious that the less overall prevalence of rare mutations not included in the screening test panel is, the more efficient the screening is. It is really important for genetic consultations to assess the residual risk at which the patient who had been screened “negative” can still be the carrier. The residual risk is defined as cumulative incidence of carriage of all pathologic variants in the examined population excluding the incidence of markers comprised in the panel [1]. Residual risk depends on the prevalence of the disease in the population, which in turn depends on ethnic features. However, for many rare diseases not all genetic defects are known, and the residual risk rate can change over time. Therefore, this method of screening panel assessment can be considered effective in ethnically homogenous population, where the prevalence of genetic markers is well-known [2]. In the same line, it is fairly hard to assess its efficacy for heterogeneous or poorly studied populations, because the prevalence of one or another genetic variant is unknown or is inaccurately identified.
An alternative method of screening diagnostic efficacy assessment can be a risk-based approach with quantitative evaluation of the residual risk. The basis of risk-based management is defined in a family of ISO 31000 standards, which were developed by the International Organization for Standardization. These standards specify the term ‘residual risk’ as the risk left over after implementation of the risk control interventions [3]. For specific screening panels such intervention is the assessment of the screening results.
General formula for residual risk calculation:
Residual risk = Inherent risks - impact of interventions for risk control.
For genetic screening, inherent risks reflect the chance of birth of an affected child with a specific disease in an examined population, and screening with a relevant panel will be an appropriate intervention for risk control. The detection of carriage is assumed to almost completely exclude the risk of the birth of an affected child; in this case the residual risk will be associated with the disease genetic markers non-detectable by the test panel, defining its ‘vulnerability’*, as specified in the article 3.6.1.6 of ISO guide 73:2009 [3] (*‘vulnerability’ – intrinsic properties or weak points resulting in the object’s susceptibility to a risk source, that can lead to an event with an outcome).
Model disease
To conduct the study we chose a screening panel for cystic fibrosis screening in Russian population. In Russia the average incidence of cystic fibrosis is 1:10,000 [4] and may vary from region to region; for example, in Chuvash Republic the indicator is 4,143 in 10,000 children, while in Kabardino-Balkaria Republic it is 0,462 in 10,000. [5]. Cystic fibrosis is a systemic inherited disease due to mutation in transmembrane conductance regulator gene (CFTR-gene) [6] that leads to exocrine gland impairment and manifests with serious respiratory, gastro-intestinal tract and pancreas disorders, and with male infertility. Identification of mutations in CFTR in perspective parents in combination with assisted reproductive technologies allows preventing the birth of an affected child. Such practice is widely spread in some countries. For instance, in USA CFTR mutation carrier testing is offered to all patients of reproductive age regardless of family history. This carrier screening strategy was recommended by the American College of Medical Genetics and Genomics (ACMG) [7] and the American College of Obstetricians and Gynecologists (ACOG) [8]. At the same time, the European Cystic Fibrosis Society (ECFS) leaves the decision on carrier screening to the discretion of the European countries themselves [9]. Cystic fibrosis carrier screening is offered in Israel [10], some districts of Australia [11], and in Italy [12]. Carrier testing for cystic fibrosis has been found to be associated with decreased annual incidence of the disease in the United Kingdom, the USA and in Italy [12–14].
Since 2006, as a part of the ''Health'' National Project, neonatal screening has been conducted in Russia to detect severe hereditary diseases, including cystic fibrosis, phenylketonuria, hypothyroidism, galactosemia and congenital adrenal hyperplasia [15].
In Russia, the screening protocol for cystic fibrosis consists of 4-staged sequential testing: test for immunoreactive trypsinogen (IRT) in blood, repeated IRT test, sweat test (conducted in case of IRT exceeding the threshold) and DNA diagnostics [16]. Nowadays only the first three steps are mandatory, DNA diagnostics is mostly performed for ambiguous results or when it is not possible to perform a sweat test. Nevertheless, the severity of the disease is strongly associated with the identification of the class of mutations [17], and genotype confirmation allows recommending the patients to try a targeted pharmacogenetic therapy [17, 18].
According to the neonatal screening regulations in the Russian Federation, molecular genetic testing for cystic fibrosis includes several steps. The first is a search for frequent mutations using specific screening panels that contain the most frequent mutations [16, 17, 19]. If frequent mutations are not identified, then a gene sequencing is performed [17, 20].
To date there are no carrier screening programs in Russia which allow detecting the most frequent mutations associated with cystic fibrosis, though there are various test panels on the market. However, the value of these panels for reduction of the risk of birth of an affected child has not been assessed yet. Therefore, the aim of this study was to evaluate the effectiveness of a screening panel for carriage of CFTR gene mutations in Russia using a risk-based approach.
Materials and methods
Experimental assessment of the frequency of mutation carriage was performed using genotyping of 1,000 healthy blood donors, living in Russia. Blood samples were obtained as a part of the research project «Distribution of clinically relevant genetic markers (certain nucleotides) in the Russian population» (2012–2014), carried out in Petrovsky National Research Centre of Surgery and the Institute of Immunology of the Federal Medical-Biological Agency (Ethical Committee Ref. No. 8 of September 5, 2012). All participants signed an informed consent to participate in the study. All patients’ information was depersonalized.
DNA purification from 0.1 ml peripheral blood was performed with the use of «PROBA-GS-GENETIKA» kit by «DNA-Technology» (Moscow, Russia). The DNA samples were immediately genotyped or were preserved at -20°С. Single nucleotide substitutions were identified using Real Time PCR in combination with the analysis of melting profile and a special kit “Genetics of hereditary diseases. Cystic fibrosis screening”, “Genetics of hereditary diseases. Cystic fibrosis – rare mutations”. These kits allow the detection of 24 mutations in CFTR-gene associated with the development of cystic fibrosis.
According to research literature, F508del mutation is found in 51,67% of cases, dele2,3(21kb) – 5,68%, E92K – 2,43%, 3849+10kbC>T – 2,1%, 2143delT – 1,9%, W1282X – 1,82%, 2184insA – 1,8%, N1303K – 1,35%, G542X – 1,18%, L138ins – 1,07%, 1677delTA – 0,95%, 394delTT – 0,82%, R334W – 0,8%, 3821delT – 0,45%, S1196X – 0,33%, 2789+5G>A – 0,33%, 3944delGT – 0,29%, R553X – 0,18%, 621+1G>T – 0,16%, R117H – 0,06%, 604insA – 0,06%, 2183AA>G – 0,04%, K598ins – 0,02%, 3667insTCAA – 0,01%. Cumulative mutation rate within the panel (informative value) is 75,5% [5, 21].
For confirmation, selective automatic Sanger sequencing was performed using an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, USA), reagents and manufacturer’s recommendations.
Results
Real Time PCR sequencing produced in all cases identical results of genotyping.
Routinely, the proportion of undetectable mutations among all mutations associated with the disease is calculated for carrier screening panels as follows:
V=q-qi (1), where
V is the prevalence of undetectable mutations;
q is the prevalence of all mutations (pathological allele);
qi is the prevalence of mutations detectable by the panel in the population.
Basically V is a quantitative characteristic of the panel vulnerability. According to the Hardy-Weinberg equation,
p2+2pq+q2=1 (2), where
р is the prevalence of a normal allele.
Respectively, for autosomal recessive disease with high penetrance, the prevalence of the disease (Р) will correspond to the population frequency of homozygous individuals:
P=q2 (3), where
Р is the prevalence of the disease.
Depending on (1)
Р=(V+qi)2 (4).
The prevalence of heterozygous disease markers carriage in the population from equations (2) and (1) is calculated as follows:
H=2p(V+qi) (5), where
Н – the prevalence of heterozygous disease markers carriage in the population.
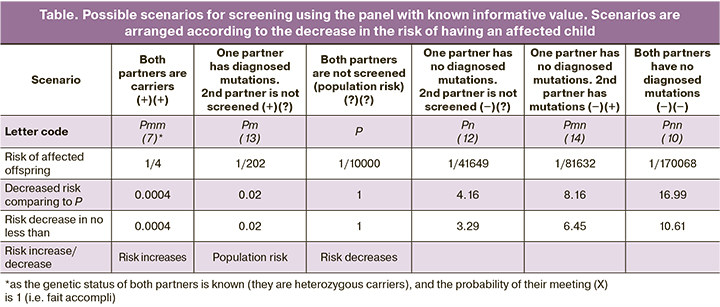
Autosomal recessive disease inheritance (not to mention de novo mutations, which are rather rare) means the birth of an affected child to a couple where both partners are heterozygous carriers of genetic disease markers. Since for most of rare genetic diseases there is no assortative mating, a probability of mating between two heterozygous carriers in the population is a compound probability of two independent events. According to the probability theory, the probability of simultaneous occurrence of two independent events is the product of their probabilities. Therefore, the probability of the event in which both partners will be heterozygous carriers is
Х= H2 = 4p2(V+qi)2 (6), where
Х is the probability in which both partners in a random couple will be heterozygous carriers of the disease markers.
The prevalence of a disease (P) will depend on the probability of mating of two heterozygous carriers (Х) and the probability of birthing an affected child in this couple. As it follows from the law of independent assortment, the probability of having an affected child in two heterozygous carriers is 0.25:
P=0.25X=0,25H2 = p2(V+qi)2 (7).
If the patient was examined, and known mutations were excluded, then qi=0, and the probability of heterozygous carriage (Hn) will be substantiated only by the probability of an unknown mutations carriage:
Hn=2p(V) (8).
In this case the probability of mating of two carriers of unknown mutations (Хnn) is
Хnn=Hn2= 4(p〖V)〗2 =4p2V2 (9),
and the probability of having an affected child is
Pnn=0.25Xnn = p2V2 (10)
If only one partner was examined (and known mutations were excluded), the probability that he/she is a heterozygous disease marker carrier (Hn) will depend on the prevalence of undetectable markers (8), and for the second partner – on the prevalence of all markers, since his/her genetic status is unknown. The probability that both partners are heterozygous carriers of the disease (Xn) is the product of the following probabilities:
Хn= H×Hn=2p(V+qi)×2p(V)=4p2V(V+qi) (11),
and the probability of affected offspring is
Pn=0.25×Xn= p2V(V+qi) (12).
If heterozygous carriage of a pathological allele in one of two partners (the probability of carriage is 1) was identified, then the risk of affected offspring in case of an unknown status of the second partner is
Pm=0.25×1×H=0.25×2p(V+qi) (13).
If the carriage of known mutations in the second partner is excluded, then this risk is
Pmn= 0.25×1×2p(V) (14).
When heterozygous disease markers carriage is identified in both partners, the risk of affected offspring (Pmm) is 0.25, as it follows from the law of independent assortment.
These formulas for calculating the residual risk of having an affected child (10, 12–14) are helpful for population studies, since the prevalence of the disease for most of populations is well known [5, 8], and there is no need for extra diagnostic methods to detect mutations not included in the screening panel.
In the present study, according to the results of genotyping of 1,000 healthy donors by 24 frequent mutations, 29 carriers of CFTR mutations were found. Cumulative prevalence of the detectable mutations was 2.9%, which is considered to be rather high in comparison with expected indicators and findings by other researchers [4, 22]. F508del mutation carriage was identified in 15 cases, R117H – in 4, N1303K – in 3, 3849+10kbC>T – in 3, dele2,3(21kb) – in 1, E92K –in 1, L138ins – in 1 and K598ins – in 1 case.
Since the study was conducted with the selected data, the projection of its results on total population should contain error element of random sampling. Confidence interval (CI) is a precision measure of an estimated variable. Thus, the true prevalence of the mutations identified by the screening panel may be found within the range from qimin to qimax.
To calculate CI of a sign’s part, the Wald's maximin model is most often used, though it is associated with significant limitations. This model is not recommended for small samples, as well as in cases when the sign prevalence approaches 0 or 1 (less than 25%, or more than 75%). The model cannot be used when the sign prevalence is 0 or 1 [23]. Wilson formula, which is more universal, allows calculating CI for very small and very large prevalence and also applies to small samples. For this study, CI (qimin - qimax), which is the prevalence of detectable carriers within the population, calculated according to the Wilson formula, was 2.0-4.1%.
The greater the vulnerability of the screening panel, the smaller is the number of mutations it can identify (1), so we must consider the worst-case scenario from a risk management perspective, i.e. the lower threshold of CI for the frequency of detected carriers (qimin), which conforms the maximum vulnerability. In this case vulnerability of the proposed panel will be no more than 100-(2.0/2.9)=31% (95% CI). The derived estimate is sufficiently close to vulnerability, theoretically calculated from the incidence of the included mutations (24.5%).
It should be noted that the limits of 95% CI are not symmetric, and the greater the asymmetry, the closer to the edge of the scale the estimated value of the part is. In our case the upper value error for the frequency of the detected carriers (which conforms minimum vulnerability) is much bigger than the lower value error (which conforms maximum vulnerability). That is why the panel vulnerability, calculated using upper value (qimax), will be less than 0, which is impossible because of the physical significance of this value. Therefore, the proposed method that shows how to assess vulnerability of the screening test panel may be used only for the upper CI threshold value. It is sufficient for risk management and can be utilized to conduct comparative studies like “not worse than”.
According to the data on the frequency of carriage (29/1,000=0.029) and informative value of the panel (0.755), the residual risk of carriage after screening will be 0.029×(1-0.755)=0.007105 ≈ 1/140, and the risk calculated using a risk-based approach, corrected to uncertainty, will be no more than 0.029×0.31 ≈ 1/112 .
Besides, owing to the above mentioned formulas, it is possible to calculate the risk of affected offspring in terms of all possible screening scenarios for both partners (see the Table).
Conclusion
We found 29 mutation carriers in 1,000 healthy individuals screened for 24 frequent mutations in CFTR gene. According to the literature, cumulative frequency of mutations in the panel used for the study (informative value) was 75.5%. In case there is no mutations in CFTR gene in both parents, use of this panel helps to reduce the risk of affected offspring by 17 times (1/170,068), comparing to the general population risk.
Risk-based approach allowed us to find out that the vulnerability of the screening panel with 95% CI does not exceed 31%, which is rather close to the theoretically calculated vulnerability that depends on the prevalence of included mutations. Such approach to the assessment of screening panels efficacy may be used in any mixed population, where the morbidity is known; this is especially relevant for the Russian Federation.
The assessment of screening panel vulnerability is an important part of the Cost Effective Approach analysis and will help to validate the advisability of genetic screening for various autosomal recessive diseases.
References
- Gregg A.R. Expanded carrier screening. Obstet. Gynecol. Clin. North Am. 2018; 45(1): 103-12. https://dx.doi.org/10.1016/j.ogc.2017.10.005.
- Scott S.A., Edelmann L. Experience with carrier screening and prenatal diagnosis for 16 Ashkenazi Jewish genetic diseases. Hum. Mutat. 2010; 31(11): 1240-50. https://dx.doi.org/ 10.1002/humu.21327.
- ГОСТ Р 51897-2011/Руководство ИСО 73:2009 Национальный стандарт Российской Федерации. Менеджмент риска. Термины и определения. [ISO GUIDE 73:2009 Risk management. Vocabulary (in Russian)].
- Капранов Н.И., Каширская Н.Ю., ред. Муковисцидоз. M.: Медпрактика-М; 2014. 672 с. [Kapranov N.I., Kashirskaya N.Yu., ed. Cystic fibrosis. M.: Medpraktika-M; 2014. 672p. (in Russian)].
- Воронкова А.Ю., Амелина Е.Л., Каширская Н.Ю., Кондратьева Е.И., Красовский С.А., Старинова М.А., Капранов Н.И., ред. Регистр больных муковисцидозом в Российской Федерации. 2017 год. М.: Медпрактика-М; 2019. 68 с. [Voronkova A.Yu., Amelina E.L., Kashirskaya N.Yu., Kondratieva E.I., Krasovsky S.A., Starinova M.A., Kapranov N.I., ed. Register of patients with cystic fibrosis in the Russian Federation. 2017. M.: Medpraktika-M; 2019, 68 p. (in Russian)].
- Kerem B., Rommens J.M., Buchanan J.A., Markiewicz D., Cox T.K., Chakravarti A. et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989; 245(4922): 1073-80. https://dx.doi.org/10.1126/science.2570460.
- Watson M.S., Cutting G.R., Desnick R.J., Driscoll D.A., Klinger K., Mennuti M. et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet. Med. 2004; 6(5): 387-91. https://dx.doi.org/ 10.1097/01.gim.0000139506.11694.7c.
- American College of Obstetricians and Gynecologists Committee on Genetics. ACOG Committee Opinion No. 486: Update on carrier screening for cystic fibrosis. Obstet. Gynecol. 2011; 117(4): 1028-31. https://dx.doi.org/10.1097/AOG.0b013e31821922c2.
- Castellani C., Macek M. Jr, Cassiman J.J., Duff A., Massie J., ten Kate L.P. et al. Benchmarks for cystic fibrosis carrier screening: a European consensus document. J. Cyst. Fibros. 2010; 9(3): 165-78. https://dx.doi.org/10.1016/j.jcf.2010.02.005.
- Zlotogora J. Population programs for the detection of couples at risk for severe monogenic genetic diseases. Hum. Genet. 2009; 126(2): 247-53. https://dx.doi.org/10.1007/s00439-009-0669-y.
- Massie J., Petrou V., Forbes R., Curnow L., Ioannou L., Dusart D. et al. Population-based carrier screening for cystic fibrosis in Victoria: the first three years experience. Aust. N. Z. J. Obstet. Gynaecol. 2009; 49(5): 484-9. https://dx.doi.org/10.1111/j.1479-828X.2009.01045.x.
- Castellani C., Picci L., Tamanini A., Girardi P., Rizzotti P., Assael B.M. Association between carrier screening and incidence of cystic fibrosis. JAMA. 2009; 302(23): 2573-9. https://dx.doi.org/10.1001/jama.2009.1758.
- Cunningham S., Marshall T. Influence of five years of antenatal screening on the paediatric cystic fibrosis population in one region. Arch. Dis. Child. 1998; 78: 345-8.
- Witt D., Wold C., Goonewardena P., Louie E., Rosenfeld S. Cystic fibrosis prenatal screening of 103,600 individuals in an HMO: molecular/clinical outcomes and a dramatic reduction in CF incidence. Proceedings of Annual Meeting, American Society of Human Genetics. Philadelphia, PA, USA; Nov 11-15, 2008. 686 (abstr.).
- Министерство здравоохранения и социального развития Российской Федерации. Приказ от 22.03.2006 № 185 «О массовом обследовании новорожденных детей на наследственные заболевания». [Ministry of Health and Social Development of the Russian Federation. Order No. 185 dated 22.03.2006 "On mass examination of newborn children for hereditary diseases". (in Russian)].
- Шерман В.Д., Каширская Н.Ю., Капранов Н.И. Современный алгоритм диагностики муковисцидоза. Педиатрия. Журнал им. Г.Н. Сперанского. 2014; 93(4): 68-74. [Sherman V.D., Kashirskaya N.Yu., Kapranov N.I. Modern algorithm for the diagnosis of cystic fibrosis. Pediatrics. The journal named after G.N. Speransky. 2014; 93(4): 68-74. (in Russian)].
- Баранов А.А., Капранов Н.И., Каширская Н.Ю., Намазова-Баранова Л.С., Шерман В.Д., Симонова О.И., Томилова А.Ю., Савостьянов К.В., Пушков А.А., Владыкин А.Л., Шатохин Н.В. Проблемы диагностики муковисцидоза и пути их решения в России. Педиатрическая фармакология. 2014; 11(6): 16-23. [Baranov A.A., Kapranov N.I., Kashirskaya N.Y., Namazova-Baranova L.S., Sherman V.D. et al. Diagnostic Problems of Mucoviscidosis and Ways of Solution in Russia. Pediatric pharmacology. 2014; 11(6): 16-23 (in Russian)].
- Dodge J.A. A millennial view of cystic fibrosis. Dev. Period. Med. 2015; 19 (1): 9-13.
- Степанова А.А., Красовский С.А., Поляков А.В. Информативность поиска 19 частых мутаций в гене CFTR у российских больных муковисцидозом и расчетная частота заболевания в Российской Федерации. Генетика. 2016; 52(2): 231-41. [Stepanova A.A., Polyakov A.V., Krasovsky S.A. Reliability of the search for 19 common mutations in the CFTR gene in russian cystic fibrosis patients and the calculated frequency of the disease in Russian Federation. Russian Journal of Genetics. 2016; 52(2): 231-41 (in Russian)].
- Симакова Т.С., Брагин А.Г., Глушкова М.А., Петрова Н.В., Поляков А.В., Кондратьева Е.И., Шерман В.Д., Павлов А.Е. Опыт применения таргетного секвенирования для молекулярной диагностики муковисцидоза. Клиническая лабораторная диагностика. 2017; 62(5): 305-9. [Simakova T.S., Bragin A.G., Glushkova M.A., Petrova N.V., Polyakov A.V., Kondratieva E.I., Sherman V.D., Pavlov A.E. The experience of application of target sequencing in molecular diagnostic of mucoviscidosis. Klinicheskaya Laboratornaya Diagnostika/ Russian Clinical Laboratory Diagnostics. 2017; 62(5): 305-9. (in Russian)]. https://dx.doi.org/10.18821/0869-2084-2017-62-5-3-5-309.
- Литвинова М.М., Дадали Е.Л., Шевченко К.Г., Поляков А.В., Исаев А.А. Результаты генетического скрининга новорожденных на наличие наиболее частых наследственных заболеваний с аутосомно-рецессивным типом наследования. Клеточная трансплантология и тканевая инженерия. 2013; 8(3): 39-40. [Litvinova M.M., Dadali E.L., Shevchenko K.G., Poljakov A.V., Isaev A.A. Newborns genetic screening results for the most frequent autosomal recessive hereditary disorders. Kletochnaja transplantologija i tkanevaja inzhenerija/ Cell transplantation and tissue engineering. 2013; 8(3): 39-40. (in Russian)].
- Гржибовский А.М., Иванов С.В., Горбатова М.А. Анализ номинальных и ранговых переменных данных с использованием программного обеспечения Statistica и SPSS. Наука и здравоохранение. 2016; 6. 5-39. [Grzhibovski A.M., Ivanov S.V., Gorbatova M.A. Analysis of nominal and ordinal data using Statistica and SPSS Software. Nauka i Zdravoohranenie/ Science and Healthcare. 2016; 6. С: 5-39. (in Russian)].
Received 10.03.2022
Accepted 24.03.2022
About the Authors
Gaukhar Yu. Zobkova, PhD student at the Department of Medical Genetics, RMACPE, Ministry of Health of Russia, zobkova.dna@gmail.com,https://orcid.org/0000-0002-9624-0484, ResearcherID: W-8153-2019, 119049, Russia, Moscow, per. 4th Dobryninsky, 1/9.
Andrew E. Donnikov, Ph.D., Head of the Laboratory of Molecular Genetic Methods, V.I. Kulakov NMRC for OGP, Ministry of Health of Russia, a_donnikov@oparina4.ru, https://orcid.org/0000-0003-3504-2406, ReasearcherID: E-7178-2015, ScopusID: 6505485697, 117997, Russia, Moscow, Academician Oparin str., 4.
Alexander N. Prytkov, Ph.D., Associate Professor, Senior Researcher at the Department of Medical Genetics, RMACPE, Ministry of Health of Russia,
119049, Russia, Moscow, per. 4th Dobryninsky, 1/9.
Natalia S. Demikova, Dr. Med. Sci., Professor, Head of the Department of Medical Genetics, RMACPE, Ministry of Health of Russia,
119049, Russia, Moscow, per. 4th Dobryninsky, 1/9.
Aligeidar A. Ragimov, Dr. Med. Sci., Professor, Head of the Department of Intensive Care and Anesthesiology, I.M. Sechenov First MSMU, Ministry of Health of Russia (Sechenov University), ra50@mail.ru, 119991, Russia, Moscow, Bolshaya Pirogovskaya str., 2-4.
Authors’ contributions: Zobkova G.Yu. – material collection and data processing, text composition; Donnikov A.E. – concept and design of the research, data processing; Prytkov A.N. – data processing and editing; Demikova N.S. – concept and design of the research, data editing; A.A. Ragimov A.A. – biomaterial collection.
Conflicts of interest: The authors declare no conflicts of interest.
Funding: The study was performed in the framework of development and validation of screening test panel for detection of mutations associated with cystic fibrosis, phenylketonuria, galactosemia and sensorinerual nonsyndromic hearing loss with the help of real-time PCR (MonogeneScreen).
Ethical Approval: The study was approved by the Ethical Committee, Institute of Immunology of the Federal Medical-Biological Agency (Ref. No. 8 of September 5, 2012)
Authors' Data Sharing Statement: The data supporting the findings of this study are available on request from the corresponding author after approval from the principal investigator.
For citation: Zobkova G.Yu., Donnikov A.E., Prytkov A.N., Demikova N.S., Ragimov A.A.
Risk-based analysis of test panels for carrier screening for monogenic disorders.
Akusherstvo i Ginekologiya/Obstetrics and Gynecology. 2022; 4: 148-154 (in Russian)
https://dx.doi.org/10.18565/aig.2022.4.148-154
Similar Articles

