

Orenetide for the treatment of hypoactive sexual desire disorder: results of a randomized placebo-controlled trial
Hypoactive desire disorder (HSDD) is characterized by a distressing and prolonged deficiency of sexual desire. Currently, despite the high prevalence of HSDD in women, the potential of drug treatment for this problem is limited: there are only two pharmacologic treatments both with modest efficacy, approved in a limited number of countries. Desirix (INN Orenetide) – is an innovative peptide drug developed in Russia to restore sexual desire and concomitant functions in women with HSDD. Aim: This study aimed to confirm the safety and efficacy of Desirix (INN Orenetide) in premenopausal women with HSDD. Materials and methods: This randomized, double-blinded, placebo-controlled, multicenter, 12-week Phase III clinical trial in parallel arms was conducted at 19 sites within the Russian Federation (Russian CTA #688 dated 25.12.2017; ClinicalTrials.gov NCT03463707). The study enrolled 189 premenopausal women with diagnosed Lack or loss of sexual desire (ICD-10 code F52.0; equivalent to acquired generalized HSDD according to DSM-IV), aged 21–50 years. Participants were eligible if they had a regular menstrual cycle and low sexual desire for at least 24 weeks prior to screening. After 4 weeks of baseline assessment (screening period) participants were randomly assigned to receive Desirix (Orenetide, development code BP101) 2 mg/day nasal spray or identical placebo. 95/189 (50,3%) participants were randomized to Desirix (Orenetide) treatment arm, and 94/189 (49,7%) to placebo arm. After 4-weeks daily treatment period there was 8-weeks no-treatment follow-up. The primary efficacy outcome was the change in number of satisfying sexual events (SSEs) after 4 weeks of therapy compared to a baseline period. Secondary efficacy outcomes included changes in the number of SSEs in the follow-up period; changes in the number of sexual events leading to orgasm, total Female Sexual Function Index (FSFI) score, FSFI “desire” domain, total Female Sexual Distress Scale-Revised (FSDS-R) score, FSDS Item 13 (“[How often… you were] Bothered by low sexual desire?”) score and assessment of Patient Global Impression of Improvement, PGI-I) after treatment, and after 4 and 8 weeks of follow-up. The incidence and nature of adverse events (AEs) was recorded. Results: The primary efficacy endpoint was met as Desirix (Orenetide) had a higher mean change in the number of SSEs after 4 weeks of treatment with the mean treatment difference of 1,83 (95,9% CI: 0,28–3,39; р=0,02) compared to placebo. Desirix (Orenetide) significantly increased SSEs compared to placebo at the 4-week – 2,38 (95% CI: 0,62–4,13; р<0,01) and 8 week – 2,37 (95% CI: 0,70–4,05; р<0,01) follow-up visits. Greater changes in total FSFI score and for “desire”, “arousal” and “orgasm” domains, as well as PGI-I score were reported at all time-points in the Desirix (Orenetide) treatment arm (p<0,05 vs placebo). Desirix (Orenetide) effectively reduced the signs of distress on the FSDS-R scale, and increased in the mean number of sexual events leading to orgasm at 4 weeks of treatment and at 8 weeks of follow-up (p<0.05 vs placebo). Most AEs were mild, caused no trial discontinuations and no serious treatment-related AEs were reported. Conclusion: Desirix (Orenetide) medicine is an efficacious and safe treatment for HSDD in premenopausal women.Sukhikh G.T., Smulevich A.B., Stenyaeva N.N., Nemenov D.G., Prilepskaya V.N., Khritinin D.F., Myasoedov N.F., Matskevich A.A., Andreeva L.A., E.V. Zelenina E.V.
Keywords
hypoactive sexual desire disorder
lack or loss of sexual desire
Desirix
Orenetide
BP101
premenopausal women
Hypoactive sexual desire disorder (HSDD) is a distressing and persistent or recurrent deficiency of a desire for sexual activity, and absence of sexual or erotic thoughts or fantasies [1]. HSDD is a common multifactorial sexual disorder experienced by 7.4% of women in developed countries [2, 3]. Low sexual desire can be caused by an imbalance of excitatory and inhibitory pathways and neurotransmitters that regulate the sexual response [3, 4]. It is hypothesized that women with low sexual desire might exhibit an overabundance of inhibitory neuromodulators (serotonin, opioids, endocannabinoids) relative to levels of excitatory neuromodulators (norepinephrine, oxytocin, dopamine, melanocortins), however, the mechanism of a pathological decrease of sexual desire in women is currently not well understood [5].
Sexual dysfunction may have a profound adverse effect on women’s overall health and on their relationships with partners and loved ones [3]. In a large survey that examined the effects of sexual dysfunction on quality of life, 70% of women with suboptimal sexual desire identified personal and interpersonal consequences. These included loss of connectedness with a sexual partner and negative self perception in body image and confidence[5,6]. HSDD often remains untreated [7]. Currently, flibanserin and bremelanotide are the only approved drugs for HSDD treatment in premenopausal women, however, they are officially sold only in the USA [8, 9]. Both flibanserin and bremelanotide have difficulties in product usage and modest efficacy [7]. There is an unmet medical need for a convenient, safe, and effective medicine for the treatment of premenopausal women experiencing HSDD.
Desirix (INN orenetide, code name BP101) is an investigational drug consisting of a five amino acid peptide, intended for bilateral intranasal spray application. The mechanism of action of Desirix is currently not fully elucidated, but there is evidence to indicate it works by acting on the brain structures responsible for the regulation of sexual and reproductive behavior [10]. Intranasal administration of Desirix stimulated sexual behavior in rats, most likely by acting on the nuclei of the hypothalamus and limbic-reticular system [10]. It has been shown that Desirix is delivered to the site of activity via the nasal mucosa to the neurons of the olfactory bulbs and then through their axons that comprise the olfactory tracts on the basal surface of the brain, into the hypothalamic structures. In vitro Desirix at high concentrations inhibits the activation of selected gamma-amino butyric acid (GABA)A receptors. These data are aligned with the established notion that GABA affects sexual behavior [11–14]. Additionally, Desirix stimulates sexual behavior in animals when injected intracranially into the preoptic area, an area with demonstrated roles in reproduction and sexual behavior [10, 15–17]. Recent data suggest that Desirix affects the mechanisms of glutamate neuronal transmission in the diencephalon and possibly in the prefrontal cortex.
Human sexuality and sexual desire are lifelong considerations in overall health and sexual activity, that remains important to mеn and women [18]. The drug "Desirix" was studied in several clinical studies: and it has been shown as safe in Phase I clinical trials BP101-HV01 and BP-101-HV02. Results from a randomized, double-blinded, placebo-controlled Phase II study of Desirix (BP-101-SD01) demonstrated that Desirix administered via nasal spray, safely and significantly improved sexual desire and sexual function and decreased associated distress in women [19].
The aim of this Phase III study was to confirm the safety and efficacy of Desirix in premenopausal women with HSDD. The study was approved by the Ministry of Health of the Russian Federation (RCT No. 688 dated December 25, 2017 in the State Register of Medicines grls.rosminzdrav.ru/CIPermitionReg.aspx) and independent Ethics Committees of each of the participating clinical centers. Before being included in the study, all participants provided written informed consent to participate in it. Information about the study was entered into the international register of clinical trials at ClinicalTrials.gov, with the number NCT03463707.
Materials and methods
Study design
This randomized, double-blinded, placebo-controlled, multicenter, 12-week trial in parallel arms was conducted in women with HSDD at 19 outpatient sites in the Russian Federation from March 2018 to February 2019. At baseline and further during the study, participants filled in an electronic diary for 28–30 days to assess their baseline sexual function status. Also at baseline participants underwent laboratory and instrumental screening tests and assessments using relevant questionnaires and scales. After the end of the screening period and final assessment of all eligibility criteria, the participants were randomized to receive treatment with either Desirix or placebo in a 1:1 ratio.
The course of the therapy lasted for 4 weeks, after the completion of treatment, the participants were observed for another 8 weeks, during which the participants did not receive any treatment.
During this study, one amendment to the Protocol was issued, which was previously approved by the Ministry of Health of the Russian Federation and all independent Ethics Committees. The changes introduced by the amendment addressed identified internal inconsistencies and minor protocol errors; the scheduled timing of the interim analysis was also postponed: this analysis was scheduled after the inclusion of 189 randomized participants instead of 160 as originally planned. The shift in the scheduled timing of the interim analysis was due to the specific technical features of the recruitment of participants in this study. The changes made did not affect the design of the study, the qualitative composition of the participants, the scope and results of the evaluation procedures.
Study participants and key study entry criteria
The inclusion criteria for the study were: an established diagnosis of a decrease or loss of sexual desire (ICD-10 code: F52.0); corresponding to diagnosis of HSDD according to the Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV criteria, with a current episode lasting longer than 6 months; age of women from 21 to 50 years; stable, monogamous, heterosexual relationships for at least 1 year; a regular menstrual cycle. Premenopausal status was defined as Staging System for Reproductive Aging in Women (STRAW) stages -5 to -3. Participants agreed, in case of such a desire appeared, to attempt sexual intercourse at least 2 times a month, with a partner physically available, for at least 50% of the time each month, while using effective contraceptive methods throughout the study. To be eligible, participants had to have a minimum score of 15 on the Female Sexual Distress Scale-Revised (FSDSR) Total Score. Participants were required to complete the electronic diary for at least 80% of the days during the baseline assessment.
Participants were excluded from this study if there was a prior primary diagnosis of another sexual dysfunction or major psychiatric disorder, or suspected depressive disorder, assessed as having a score of 20 or more on the Beck Depression Scale at screening within the past 12 months. Other exclusion criteria included participants receiving psychotherapy treatment for sexual problems and/or relationship problems with a sexual partner within the past 12 months. Participants were excluded in the case of taking drugs prohibited in the study, including antidepressants (and specifically selective serotonin reuptake inhibitors and serotonin norepinephrine reuptake inhibitors). Other exclusion criteria were: the presence of acute and exacerbation of chronic inflammatory diseases of the nasal cavity and nasopharynx, the presence of somatic diseases or other reasons that prevent regular sexual activity, alcohol dependence, severe life stressors, or drug dependence in anamnesis in the previous 12 months.
Participants were withdrawn from the study in the event of pregnancy, non-compliance with the rules for participation in the study, and also if it was found that the participant was included in the study in violation of the eligibility criteria.
Investigational drug, randomization and masking
Study participants were randomly assigned (randomized) in a 1:1 ratio to either Desirix, administered as a nasal spray at a dose of 2 mg 1 time per day (one spray puff containing 1 mg of the drug in each nasal passage), or into the placebo arm, externally completely identical to Desirix. The first use of the drug was made after training and under the supervision of medical personnel. Participants then self-administered the study drug at home. The treatment lasted for 4 weeks.
Randomization was carried out centrally using an Interactive Web Response System [IWRS] based on a pre-created computer algorithm (randomization list) with the assignment to each participant of a randomization number linked with the individual number of the vial with the study drug (block randomization was used where participants were block randomized with a block size of 4). As a result of the randomization procedure, the staff of the research center through the IWRS system received information about the number of the vial, which was dispensed to the study participant.
In this study, the possibility of influencing the choice of the arm into which a particular participant was assigned during randomization was excluded. The randomization scheme (randomization list) was not known to the study site staff, nor to the participants, nor to other personnel directly involved in the study. Thus, none of them knew what treatment a particular study participant was receiving.
Assessment methods
Safety assessments included adverse events (AEs), clinical laboratory and instrumental assessments, and vital signs measurements. Additionally, blood concentrations of the following hormones were assessed in all participants: luteinizing, follicle-stimulating, estradiol, prolactin, and total testosterone. Reports of AEs, including type and intensity, were collected throughout the study. Laboratory parameters were assessed at baseline, at the end of treatment, and at the end of each month during follow-up; body weight and vital signs were measured at every clinic visit. AEs were coded using Medical Dictionary for Regulatory Activities (version 21.1).
An electronic participant diary was used to record daily characteristics of sexual interactions including satisfying sexual events (SSEs) and sexual events leading to orgasm. Other relevant assessments were made using Female Sexual Function Index (FSFI), Female Sexual Distress Scale-Revised (FSDS-R), and Patient Global Impression of Improvement (PGI-I) scales every 4 weeks using self-assessment tools validated for the assessment of patients with HSDD. All scales and questionnaires were applied in Russian using validated translations.
Endpoints
The primary efficacy endpoint was the change in the number of SSEs after 4 weeks of therapy compared with baseline. Secondary efficacy endpoints included changes in the number of SSEs in the follow-up after completion of treatment, as well as changes in the number of sexual events leading to orgasm, FSFI total score, FSFI domains scores (“desire”, “arousal”, “lubrication”, “orgasm”, “satisfaction” and “pain” [associated with vaginal penetration]), FSDS-R total score, FSDS-R Item 13 score, and PGI-I scale score after 4 weeks of treatment, and after 4 and 8 weeks of no-treatment follow-up. All changes were compared between treatment arms and with baseline.
During the treatment period, participants kept a self-assessment diary recording the number of doses taken by the participant between visits. Investigators monitored the diaries to control participants’ compliance. The acceptable level of participant compliance was defined as the use of the product for at least 80% of the days of the planned treatment course.
Statistical analysis and sample size calculation
Safety analyzes were performed for all participants who received at least one dose of study drug or placebo. Efficacy analyzes for all endpoints were performed in the full analysis population, which included all randomized participants who received at least one dose of study drug or placebo. This population complied with a modified intention to treat analysis (mITT). The modification was to include in the analysis participants who had baseline data, and data for analysis of at least one efficacy measure after the start of the treatment. To recover missing data, the last observation carried forward (LOCF) and baseline observation carried forward (BOCF) methods were used. For performance indicators based on the participant's diary data (satisfying sexual events, number of sexual events leading to orgasm), standardization was performed for a 28-day (4-week) period using the following formula: 28 × (number of events / number of completed days in the diary).
This study tested the statistical hypothesis of superiority of Desirix over placebo in the primary endpoint – change in number of SSEs after 4 weeks of treatment compared to baseline. In connection with the pre-planned unblinded interim analysis with the possibility of stopping in case of early efficacy demonstration or with recalculation of the required number of participants, to adjust the level of significance due to the multiplicity of comparisons, a function of the dependence of the probability of a type 1 error on the amount of data obtained (alpha spending function) of the Pocock type was chosen. The value of the two-tailed alpha level for the interim analysis was set at 0.041, and for the final analysis – 0.024, respectively (with an overall two-tailed significance level in the study of 5%, one-tailed 2.5%).
Due to the completion of the study after an early demonstration of efficacy during the interim analysis, comparison results for the main endpoint are presented at a two-tailed significance level of 0.041. At the same time, for secondary endpoints, a critical value of a two-sided significance level of 0.05 (5%) was used, without adjustment.
The primary efficacy endpoint was analyzed using the analysis of covariance (ANCOVA) with the treatment as a fixed factor and the baseline value as a covariate. The same method was used to analyze the change in the number of SSEs in the no-treatment follow-up, the number of sexual events leading to orgasm, the FSFI total score and its domains, the FSDS-R total score, and separately on the Item 13 score. The results on the PGI-I scale were analyzed by the analysis of variances (ANOVA) for all visits. Validation of applicability was carried out using graphical methods, including the analysis of residuals by models.
All statistical analyses were performed using the SAS 9.4 statistical package.
The sample size was calculated based on the Phase II clinical trial data, the expected standardized effect size for the change in the number of SSEs in this study was about 0.47. Assuming this effect size with a two-sided significance level of 0,024 and an not less than 85% of the test power to detect differences between the mean values of the primary efficacy criterion in the treatment arms, , the number of participants in each treatment arm included in the final statistical analysis should have been 100 in each treatment arm (200 participants in total). Considering a possible 10% participants withdrawal rate and a factor of 1.15 due to the possible use of non-parametric analysis methods for SSE, it was recommended that at least 256 participants be randomized, 128 to each treatment arm. An interim analysis was planned in this study after τ=0.74 of the study duration (based on data from 189 randomized participants). The estimation was performed using the PASS 13 software package.
Results
Disposition of study participants
A total of 205 participants were screened for this study, of which 189 participants were randomized: 95/189 (50.3%) to the Desirix drug arm and 94/189 (49.7%) to the placebo arm; of these, 94/95 (98.9%) participants in the Desirix arm and 90/94 (95.7%) participants in the placebo arm were included in the efficacy analysis. The final disposition of participants, indicating the reason for dropping out, is shown in Figure 1.
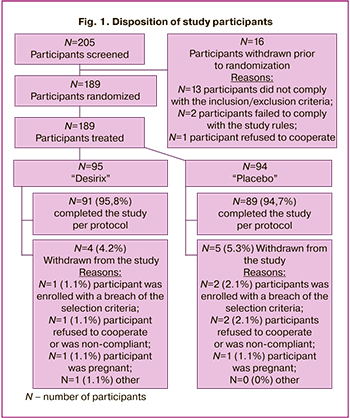
Participants were recruited, treated, and followed up between March 2018 and November 2018. A planned, unblinded interim analysis was conducted by an independent statistician based on data from 189 randomized participants and demonstrated the superiority of Desirix over placebo in the primary endpoint. Based on the results of this analysis, the independent Data Monitoring Committee recommended that the study should be prematurely completed due to the demonstration of early efficacy.
Demographic and background characteristics
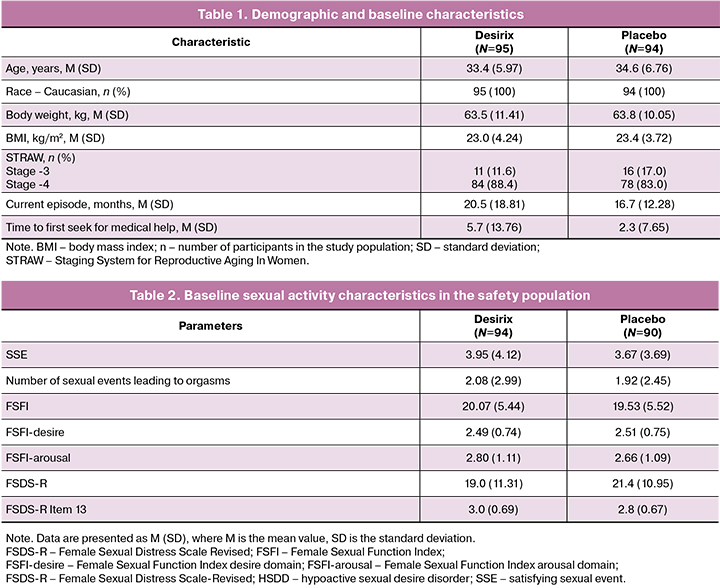
Demographic and baseline characteristics were similar between treatment arms (Table 1 and Table 2). All participants were Caucasian, their mean age was 34 (6,39) years, and their ages ranged 21–50 years. Most participants 162/189 (85.7%) were assessed at STRAW Stage -4. The mean duration of the current episode of HSDD was 20.47 (18.81) months in the Desirix arm and 16.67 (12.28) months in the placebo arm . Mean time from decrease or loss of sexual desire to the first presentation for medical care for was 5.7 (13.76) months in the Desirix arm and 2.31 (7.65) months in the placebo arm.
Safety evaluation
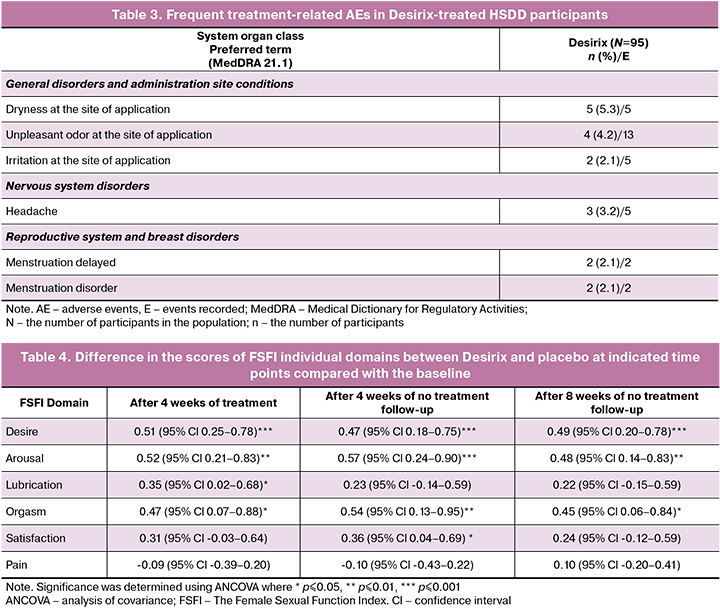
A total of 81 AEs were reported in 33 (34.7%) participants in the Desirix arm and 76 AEs in 29 (30.9%) participants in the placebo arm. Most AEs were mild (87.9%). There were no severe adverse events, and AEs requiring discontinuation of study drug or additional treatment. 1/94 (1%) participant in the placebo arm had a spontaneous abortion. The most frequent treatment-related AEs for Desirix are reported in Table 3.
Efficacy
Desirix-treated participants showed statistically significant and clinically important improvements in both the primary endpoint and majority of the secondary endpoints. The primary efficacy outcome was achieved as Desirix resulted in a greater increase in mean SSE number over 4 weeks of treatment than placebo: mean difference was 1.83 (95.9% CI: 0.28–3.39; p=0.02) (Fig. 2). Similarly, the Desirix arm also showed a significantly greater increase in SSE compared with placebo at 4 weeks 2.38 (95% CI: 0.62–4.13; p<0.01) and at 8 weeks 2.37 (95% CI: 0.70–4.05; p<0.01) of follow-up after completion of the treatment (Fig. 2).

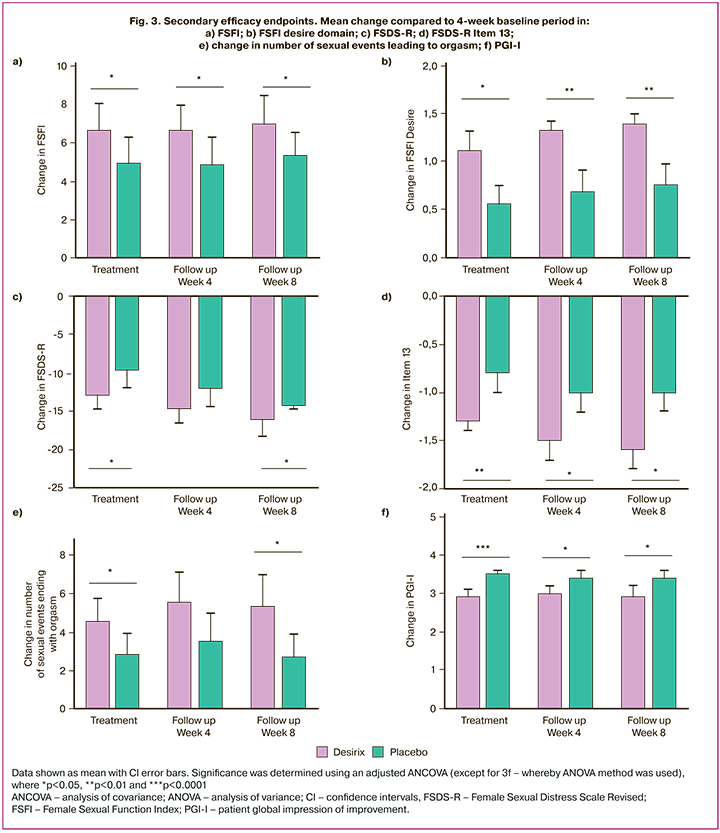
More women treated with Desirix reported a positive change in the FSFI total score with the difference between the Desirix and placebo arms was 2.03 (95% CI: 0.42–3.65; p=0.01) after 4 weeks of treatment and 2.03 (95% CI: 0.33–3.73; p=0.02) and 1.95 (95% CI: 0.24–3.66; p=0.03) at 4 and 8 weeks of follow-up after completion of treatment, respectively (Fig. 3a). The “desire”, “arousal” and “orgasm” domains of the FSFI score were significantly improved in the Desirix arm compared to placebo across all time points (Table 4). The FSFI “desire” domain was significantly improved by 0.51 (95% CI: 0.25–0.78; p<0.001) after 4 weeks of treatment, and by 0.47 (95% CI: 0.18–0.75; p<0.01) at 4 weeks and by 0.49 (95% CI: 0.20–0.78; p<0.001) at 8 weeks of follow-up after completion of the treatment (Table 4, Fig. 3b).
The difference between Desirix and placebo for FSDS R total score was -2.9 (95% CI: -5.6–0.2; p=0.04) after 4 weeks of treatment, -2.6 (95% CI: -5.5–0.3; p= 0.08) at 4 weeks and -3.6 (95% CI: -6.4–-0.8; p=0.01) at 8 weeks of follow-up after completion of the treatment (Fig. 3c). Similarly, changes in the FSDS-R Item 13 were significantly greater in the Desirix arm after 4 weeks of treatment -0.4 (95% CI: -0.7–0.1; p<0.01), and at 4 and 8 weeks of follow-up after completion of the treatment – -0.3 (95% CI: -0.6–0.0; p=0.03) and -0.5 (95% CI: -0.8–0.2 ; p<0.01), respectively (Fig. 3d).
Desirix-treated participants had significantly higher numbers of sexual events leading to orgasm after 4 weeks of treatment - 1.30 (95% CI: 0.05–2.56; p=0.04) and at 8 weeks of follow-up after completion of the treatment 1.92 (95% CI: 0.38–3.46; p=0.01) compared with the placebo arm (Fig. 3e). For the PGI-I scale the significant improvement in scores between Desirix and placebo were 0.5 (95% CI: -0.8–0.3; p<0.001), -0.4 (95% CI: -0.7–0.2; p<0.01) and - 0.5 (95% CI: -0.7–0.2; p<0.01) as assessed at weeks 4, 8, and 12, respectively (Fig. 3f).
Discussion
HSDD is a common sexual disorder in women characterized by a distressing lack of sexual desire [2, 7, 20, 21]. Despite the approval of flibanserin in 2015 and then bremelanotide in 2019, there remains an unmet need for a highly effective and safe treatment for premenopausal woman [8, 9].
This study evaluated the safety and efficacy of Desirix for the treatment of premenopausal women with HSDD. The primary efficacy endpoint was met as Desirix-treated participants had a significantly higher increase in the number of SSEs from baseline compared to the placebo. Similarly, it was demonstrated that Desirix was generally safe and well tolerated. The most common AEs experienced with Desirix treatment were headaches, nasal dryness, and an unpleasant odor. Most reported AEs were mild in severity and there were no severe AEs or AEs that led to discontinuation of the study treatment or required prescription of other medications.
Importantly, the mean change in the number of SSEs from baseline for the Desirix treatment arm was twice as high compared to placebo, thus demonstrating clinical efficacy of the treatment. Additionally, efficacy secondary outcomes was confirmed, as Desirix significantly increased number of SSEs at 4 and 8 weeks of the follow up compared with the baseline, and Desirix also demonstrated superiority over placebo for the change of the FSFI total score at all time points. The ”desire”, ”arousal” and ”orgasm” domains of the FSFI were significantly improved at all time points while superiority for “lubrication” and “satisfaction” domains only significant at the 4-week of follow-up. Also, Desirix treatment resulted in a reduction in distress as measured by the FSDS-R Item 13 score at all time points measured, and an increase in the number of sexual events leading to orgasm compared to the baseline period during treatment and 8 weeks after completion of the treatment.
The efficacy measurements used in this study have been used extensively in other clinical studies for HSDD [21–23]. Discussions have been raised about the use of change in SSEs as a primary endpoint, which has been reflected in FDA guidelines for pivotal studies in the female sexual dysfunction [23]. The SSEs assessment is based on frequency counts and arguably does not address whether sexual desire is actually increased [23]. Indeed, the current evidence suggests that well validated questionnaires, such as the FSFI domain scores, might be superior to event logs for assessing change in female sexual dysfunction in clinical trials [23]. In addition to the FSFI-D, the FSFI-total and other subdomains and the FSDS-R and FSDS-R item 13 are commonly used as outcome variables for HSDD treatment clinical trials [22, 24, 25]. This study included all these assessments to provide comprehensive data regarding the efficacy of Desirix.
In this study, 33/95 (34.7%) of participants in the Desirix arm experienced AEs. For reference, in flibanserin and bremelanotide randomized, double-blinded, placebo-controlled clinical trials AEs were reported in 56.1% and 62% of participants, respectively [26, 27]. These AEs led to 10.4% and 5% of participants treated with flibanserin and bremelanotide, respectively, been withdrawn from these clinical trials [26, 27]. In the therapeutic group of Desirix, there were no cases of discontinuation of the drug or the need to prescribe other drugs to treat emerging AEs. Additionally, there were no reported severe or serious AEs associated with Desirix treatment while both types were reported for flibanserin and bremelanotide [26,27]. A frequently reported AE in Desirix participants was headache (3.2%), and this was reported in other trials at similar levels for both flibanserin and bremelanotide [26, 27]. This suggests that Desirix may have a favorable safety profile over current HSDD therapies.
A limitation of this study is that all participants were premenopausal Caucasian women which may limit the extrapolation of the results to the general population. Additionally, it was a requirement that study participants have a stable heterosexual relationship for at least one year thus excluding other types of sexual relationships. For these reasons it is pertinent to confirm both the safety and efficacy of Desirix in a broader clinical population.
The early completion of the clinical trial based on the pre-specified condition of achievement of the primary endpoint during the interim analysis resulted in a smaller number of recruited participants. This could have led to a reduction in statistical power in the study on some of the variables and explain failure to reach statistical significance at some time-points.
Desirix was administered for 28 days which is a convenient and comparatively short treatment course. On the other hand, longer therapy may be more effective, however clinical trials with longer Desirix treatment duration are needed to confirm this.
Study limitations
This study is a multicenter, randomized, placebo-controlled in parallel arms study conducted in accordance with good clinical practice (GCP) and international guidelines. The study was conducted in a single country and included only Caucasian premenopausal women, which may limit the applicability of the results to the general population.
Conclusion
The data from this study supports the existence of a novel and highly efficient approach for a non-hormonal treatment for premenopausal women suffering from HSDD. Desirix administered intranasally was generally well-tolerated and demonstrated a statistically significant increase compared to placebo in the primary endpoint of change in the number of SSEs after 4 weeks of treatment. For all secondary efficacy criteria, the mean positive effects measured in the Desirix arm were higher versus the placebo, reaching statistically significant difference for most cases. When this is considered together with the data on the nature and frequency of AEs, Desirix has a favorable therapeutic profile and can be recommended for the treatment of premenopausal women suffering from HSDD.
References
- McCabe M.P., Sharlip I.D., Atalla E., Balon R., Fisher A.D., Laumann E. et al. Definitions of sexual dysfunctions in women and men: A consensus statement from the fourth international consultation on sexual medicine 2015. J. Sex. Med. 2016; 13(2): 135-43. https://dx.doi.org/10.1016/j.jsxm.2015.12.019.
- Rosen R.C., Connor M.K., Miyasato G., Link C., Shifren J.L., Fisher W.A. et al. Sexual desire problems in women seeking healthcare: a novel study design for ascertaining prevalence of hypoactive sexual desire disorder in clinic-based samples of U.S. women. J. Womens Health (Larchmt). 2012; 21(5): 505-15. https://dx.doi.org/10.1089/jwh.2011.3002.
- Simon J.A. Low sexual desire – is it all in her head? Pathophysiology, diagnosis, and treatment of hypoactive sexual desire disorder. Postgrad. Med. 2010; 122(6): 128-36. https://dx.doi.org/10.3810/pgm.2010.11.2230.
- Clayton A.H., Kingsberg S.A., Goldstein I. Evaluation and management of hypoactive sexual desire disorder. Sex. Med. 2018; 6(2): 59-74. https://dx.doi.org/10.1016/j.esxm.2018.01.004.
- Kingsberg S.A., Clayton A.H., Pfaus J.G. The female sexual response: current models, neurobiological underpinnings and agents currently approved or under investigation for the treatment of hypoactive sexual desire disorder. CNS Drugs. 2015; 29(11): 915-33. https://dx.doi.org/10.1007/s40263-015-0288-1.
- Sobecki J.N., Curlin F.A., Rasinski K.A., Lindau S.T. What we don’t talk about when we don’t talk about sex: results of a national survey of U.S. obstetrician/gynecologists. J. Sex. Med. 2012; 9(5): 1285-94. https://dx.doi.org/10.1111/j.1743-6109.2012.02702.x.
- Nappi R.E., Gardella B. What are the challenges in prescribing pharmacotherapy for female sexual dysfunctions? Expert Opin. Pharmacother. 2019; 20(7): 777-9. https://dx.doi.org/10.1080/14656566.2019.1582644.
- Joffe H.V., Chang C., Sewell C., Easley O., Nguyen C., Dunn S. et al. FDA approval of flibanserin – treating hypoactive sexual desire disorder. N. Engl. J. Med. 2016; 374(2): 101-4. https://dx.doi.org/10.1056/NEJMp1513686.
- Dhillon S., Keam S.J. Bremelanotide: first approval. Drugs. 2019; 79(14): 599-606. https://dx.doi.org/10.1007/s40265-019-01187-w.
- Andreev-Andrievskiy A, Lomonosov M, Popova A, Lagereva E, Clément P, Salimov R. et al. BP101 peptide promotes female sexual receptivity in the rat. J. Sex. Med. 2017; 14(3): 336-46. https://dxdoi.org/10.1016/j.jsxm.2017.01.008.
- Ågmo A., Soria P. GABAergic drugs and sexual motivation, receptivity and exploratory behaviors in the female rat. Psychopharmacology (Berlin). 1997; 129(4): 372-81. https://dx.doi.org/10.1007/s002130050203.
- Crickmore M.A., Vosshall L.B. Opposing dopaminergic and GABAergic neurons control the duration and persistence of copulation in drosophila. Cell. 2013; 155(4): :881-93. https://dx.doi.org/10.1016/j.cell.2013.09.055.
- Rodríguez-Manzo G., Canseco-Alba A. A new role for GABAergic transmission in the control of male rat sexual behavior expression. Behav. Brain Res. 2017; 320: 21-9. https://dx.doi.org/10.1016/j.bbr.2016.11.041.
- Seney M., Chang L.-C., Oh H., Wang X., Tseng G.C., Lewis D. et al. The role of genetic sex in affect regulation and expression of GABA-related genes across species. Front. Psychiatry. 2013; 4: 104. https://dx.doi.org/10.3389/fpsyt.2013.00104.
- Maejima S., Abe Y., Yamaguchi S., Musatov S., Ogawa S., Kondo Y. et al. VGF in the medial preoptic nucleus increases sexual activity following sexual arousal induction in male rats. Endocrinology. 2018; 159(12): 3993-4005.https://dx.doi.org/10.1210/en.2018-00804.
- Wei Y.C., Wang S.R., Jiao Z.L., Zhang W., Lin J.K., Li X.Y. et al. Medial preoptic area in mice is capable of mediating sexually dimorphic behaviors regardless of gender. Nat. Commun. 2018; 9(1): 279. https://dx.doi.org/10.1038/s41467-017-02648-0.
- Yamaguchi S., Abe Y., Maejima S., Tsukahara S. Sexual experience reduces neuronal activity in the central part of the medial preoptic nucleus in male rats during sexual behavior. Neurosci. Lett. 2018; 685: 155-9. https://dx.doi.org/10.1016/j.neulet.2018.08.037.
- Lindau S.T., Schumm L.P., Laumann E.O., Levinson W., O’Muircheartaigh C.A., Waite L.J. A Study of sexuality and health among older adults in the United States. N. Engl. J. Med. 2007; 357(8): 762-74. https://dx.doi.org/10.1056/NEJMoa067423.
- Nemenov D., Lomonosov M., Golikov D. 150 BP101 new molecule for HSDD treatment - results of proof-of-concept phase 2a study. J. Sex. Med. 2018;15: S41-2. https://dx/doi.org/10.1016/j.jsxm.2017.11.108.
- Shifren J.L., Monz B.U., Russo P.A., Segreti A., Johannes C.B. Sexual problems and distress in united states women: prevalence and correlates. Obstet. Gynecol. 2008; 112:(5): 970-8. https://dx.doi.org/10.1097/AOG.0b013e3181898cdb.
- Nappi P.R.E., Cucinella L., Martella S., Rossi M., Tiranini L., Martini E. Female sexual dysfunction (FSD): prevalence and impact on quality of life (QoL). Maturitas. 2016; 94: 87-91. https://dx.doi.org/10.1016/j.maturitas.2016.09.013.
- Revicki D.A., Margolis M.K., Bush E.N., DeRogatis L.R., Hanes V. Content validity of the female sexual function index (FSFI) in pre‐ and postmenopausal women with hypoactive sexual desire disorder. J. Sex. Med. 2011; 8(8): 2237-45. https://dx.doi.org/10.1111/j.1743-6109.2011.02312.x.
- Althof S.E. Outcome measurement in female sexual dysfunction clinical trials. J. Sex Marital Ther. 2005; 31(3): 153-66. https://dx.doi.org/10.1080/00926230590909989.
- Meston C.M. Validation of the Female Sexual Function Index (FSFI) in women with female orgasmic disorder and in women with hypoactive sexual desire disorder. J. Sex Marital Ther. 2003; 29(1): 39-46. https://dx.doi.org/10.1080/713847100.
- Derogatis L., Clayton A., Lewis-D’Agostino D., Wunderlich G., Fu Y. Validation of the female sexual distress scale-revised for assessing distress in women with hypoactive sexual desire disorder. J. Sex. Med. 2008; 5(2): 357-64.https://dx.doi.org/10.1111/j.1743-6109.2007.00672.x.
- Portman D.J., Brown L., Yuan J., Kissling R., Kingsberg S.A. Flibanserin in postmenopausal women with hypoactive sexual desire disorder: Results of the PLUMERIA Study. J. Sex. Med. 2017; 14(6): 834-42. https://dx.doi.org/10.1016/j.jsxm.2017.03.258.
- Clayton A.H., Althof S.E., Kingsberg S., DeRogatis L.R., Kroll R., Goldstein I. et al. Bremelanotide for female sexual dysfunctions in premenopausal women: a randomized, placebo-controlled dose-finding trial. Womens Health (London). 2016; 12(3): 325-37. https://dx.doi.org/10.2217/whe-2016-0018.
Received 21.07.2022
Accepted 05.08.2022
About the Authors
Gennadiy T. Sukhikh, Academician of the RAS, Dr. Med. Sci., Professor, Director, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, 117997, Russia, Moscow, Ac. Oparin str., 4.Anatoliy B. Smulevich, Academician of the RAS, Dr. Med. Sci., Professor, Head of the Department for the Study of Borderline Mental Pathology and Psychosomatic Disorders, MHRC, Psychiatric Hospital No. 1 named after N.A. Alexeev of the Department of Health of Moscow, 117152, Russia, Moscow, Zagorodnoe shosse, 2.
Natalia N. Stenyaeva, PhD, Senior Researcher, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, 117997, Russia, Moscow, Ac. Oparin str., 4.
Daniil G. Nemenov, M.D., Chief executive officer, IVIX Ltd., 121069, Russia, Moscow, Stolovyj lane, 6.
Vera N. Prilepskaya, Dr. Med. Sci., Professor, Head of the Scientific Polyclinic Department, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology, Ministry of Health of Russia, 117997, Russia, Moscow, Ac. Oparin str., 4.
Dmitriy F. Khritinin, Corresponding Member of the RAS, Dr. Med. Sci., Professor at the Department of Psychiatry and Narcology of the N.V. Sklifosovsky Institute of Clinical Medicine, I.M. Sechenov First Moscow State Medical University, Ministry of Health of Russia (Sechenov University), 119991, Russia, Moscow, Bolshaya Pirogovskaya str., 2-4.
Nikolay F. Myasoedov, Academician of the RAS, Dr. Chem. Sci., Professor, Head of the Laboratory of Molecular Pharmacology of Peptides, Institute of Molecular Genetics of National Research Centre «Kurchatov Institute», 123182, Russia, Moscow, Akademika Kurchatova square, 2.
Aleksey A. Matskevich, PhD (Bio), Chief Researcher, IVIX Ltd., 121069, Russia, Moscow, Stolovyj lane, 6.
Lyudmila A. Andreeva, Deputy Head of the Laboratory of Molecular Pharmacology of Peptides, Head of the Regulatory Peptides Sector of the Physiologically Active Substances Chemistry Department, Institute of Molecular Genetics of National Research Centre «Kurchatov Institute», 123182, Russia, Moscow, Akademika Kurchatova square, 2.
Elena V. Zelenina, PhD, psychiatrist, Psychiatric Hospital No. 1 named after N.A. Alexeev of the Department of Health of Moscow,
117152, Russia, Moscow, Zagorodnoe shosse, 2.
Corresponding author: Daniil G. Nemenov, d.nemenov@ovocabio.com
Authors' contributions: All authors participated in the conception, design, and implementation of the study. All authors were involved in the interpretation of analyzed data and the decision to submit for publication.
Conflicts of interest: Authors D.G. Nemenov and A.A. Matskevich are employees of IVIX Ltd.
Funding: This sudy was funded by IVIX Ltd.
Ethical Approval: This Study was approved by Ministry of Healthcare of Russian Federation (Russian CTA #688 dated 25.12.2017 in the state register of medicines grls.rosminzdrav.ru/CIPermitionReg.aspx) and by Independent Ethics Committees of each of the clinical sites participating in the study.
Patient Consent for Publication: All study participants have signed the informed consent for study participation and publication of de-personalized data, gathered in this study.
Authors' Data Sharing Statement: The data supporting the findings of this study are available on request from the corresponding author after approval from the principal investigator.
For citation: Sukhikh G.T., Smulevich A.B., Stenyaeva N.N., Nemenov D.G., Prilepskaya V.N., Khritinin D.F., Myasoedov N.F., Matskevich A.A., Andreeva L.A., Zelenina E.V.
Orenetide for the treatment of hypoactive sexual desire disorder:
results of a randomized placebo-controlled trial.
Akusherstvo i Ginekologiya/Obstetrics and Gynecology. 2022; 8: 95-106 (in Russian)
https://dx.doi.org/10.18565/aig.2022.8.95-106

